Download

1 / 45
450 likes | 658 Vues
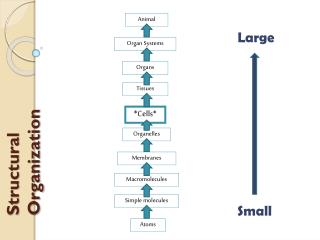
Lecture 5: Protein Structure Prediction Methods . Chen Yu Zong Tel: 6874-6877 Email: csccyz@nus.edu.sg http://xin.cz3.nus.edu.sg Room 07-24, level 7, SOC1 National University of Singapore. Protein Structural Organization. Proteins are made from just 20 kinds of amino acids.

E N D
Lecture 5: Protein Structure Prediction Methods. Chen Yu ZongTel: 6874-6877Email: csccyz@nus.edu.sghttp://xin.cz3.nus.edu.sgRoom 07-24, level 7, SOC1National University of Singapore
Protein Structural Organization Proteins are made from just 20 kinds of amino acids
Protein Structural Organization Protein has four levels of structural organization
Protein Secondary Structure Prediction: • Secondary structure forms early in protein folding process. • Identification of secondary structural elements makes the topology of protein structure more obvious—so that similar ones can be identified in a topology database such as TOPS. • Prediction of the positions and lengths of secondary structure elements can be used as a prelude to "docking" these secondary structural elements against each other • Useful guide in the construction or refinement of primary structure alignments, and to the correct correspondence between parts of two proteins' respective tertiary structures. • Useful for making some kind of intelligent guess about the higher order structure of your protein
Protein Secondary Structure Prediction: Traditional methods: CF, GOR – Accuracy 60% Recent improvements: Neural network, homologous sequences – Accuracy > 70% References: • "Prediction of the secondary structure of proteins from their amino acid sequence", P. Y. Chou, G. D. Fasman, 1978, Adv. Enzymolog. Relat. Areas Mol. Biol., 47, 45-147. • "GOR method for predicting secondary structure from amino acid sequence", J. Garnier, J.-F. Gibrat, B. Robson, 1996, Methods Enzymol., 266, 540-553. • "Analysis of the accuracy and implications simple methods for predicting the secondary structure of globular proteins", J. Garnier, D. J. Osguthorpe, B. Robson, 1978, J. Mol. Biol., 120, 45-147. • "Improvements in protein secondary structure prediction by an enhanced neural network", Kneller, 1990, J. Mol. Biol., 214, 171-182
Protein Secondary Structure Prediction: Software: • Zvelebil, M.J.J.M., Barton, G.J., Taylor, W.R. & Sternberg, M.J.E. (1987). Prediction of Protein Secondary Structure and Active Sites Using the Alignment of Homologous Sequences Journal of Molecular Biology, 195, 957-961. (ZPRED) • Rost, B. & Sander, C. (1993), Prediction of protein secondary structure at better than 70 % Accuracy, Journal of Molecular Biology, 232, 584-599. PHD) • Salamov A.A. & Solovyev V.V. (1995), Prediction of protein secondary strurcture by combining nearest-neighbor algorithms and multiply sequence alignments. Journal of Molecular Biology, 247,1 (NNSSP) • Geourjon, C. & Deleage, G. (1994), SOPM : a self optimized prediction method for protein secondary structure prediction. Protein Engineering, 7, 157-16. (SOPMA) • Solovyev V.V. & Salamov A.A. (1994) Predicting alpha-helix and beta-strand segments of globular proteins. (1994) Computer Applications in the Biosciences,10,661-669. (SSP) • Wako, H. & Blundell, T. L. (1994), Use of amino-acid environment-dependent substitution tables and conformational propensities in structure prediction from aligned sequences of homologous proteins. 2. Secondary Structures, Journal of Molecular Biology, 238, 693-708. • Mehta, P., Heringa, J. & Argos, P. (1995), A simple and fast approach to prediction of protein secondary structure from multiple aligned sequences with accuracy above 70 %. Protein Science, 4, 2517-2525. (SSPRED) • King, R.D. & Sternberg, M.J.E. (1996) Identification and application of the concepts important for accurate and reliable protein secondary structure prediction. Protein Sci,5, 2298-2310. (DSC).
Protein Secondary Structure Prediction: Types of amino acids Hydrophobic Hydrophilic, Neutral Hydrophilic, Acidic Hydrophilic, Basic
Protein Secondary Structure Prediction: Types of Secondary Structures: Alpha helix and Beta- sheet
Protein Secondary Structure Prediction: Secondary Structures: Favored Peptide Conformation
Protein Secondary Structure Prediction: Secondary Structures: Computation of structural propensity of a residue • Data derived from proteins of known structure is used to calculate 'propensities' for each amino acid type for adopting helix, sheet or turn
Protein Secondary Structure Prediction: Secondary Structures: Computation of structural propensity of a residue Three states: alpha helix, beta sheet, turn
Protein Secondary Structure Prediction: Structural propensity of amino acids Each residue is assigned to one of the three classes: • Forming residues – favor a structure • Indifferent residues • Breaking residues – stop the extension of a structure
Protein Secondary Structure Prediction: Position specific turn parameters
Protein Secondary Structure Prediction: Chou and Fasman procedure • Find helical initiation regions • Extend helices until they reach tetrapeptide breakers • Find beta initiation regions • Extend until they reach tetrapeptide breakers • Find turns • Resolve conflicts between alpha and beta Somewhat subjective … often have overlaps. Chou and Fasman suggest using additional information: • alpha-beta pattern, i.e. does this look like an b-a-b structure ??? • end probabilities – Chou and Fasman in later papers also tabulated the preferences for the residues to occur at the amino and carboxyl terminal ends of a and b structures. These can be used to resolve overlaps Chou and Fasman did not provide an explicit algorithm for this conflict resolution, relying on their expert judgment. This meant that each person’s prediction could be different. Most people are not experts. "Prediction of the secondary structure of proteins from their amino acid sequence", P. Y. Chou, G. D. Fasman, 1978, Adv. Enzymolog. Relat. Areas Mol. Biol., 47, 45-147.
Homology Modeling: Reference: • Sanchez R, Sali A. Advances in comparative protein-structure modelling. Curr Opin Struct Biol. 1997 Apr;7(2):206-14. • Krieger E, Nabuurs SB, Vriend G. Homology modeling. Methods Biochem Anal. 2003;44:509-23 • Rodriguez R, Chinea G, Lopez N, Pons T, Vriend G. Homology modeling, model and software evaluation: three related resources. Bioinformatics. 1998;14(6):523-8 • Alexandrov NN, Luethy R. Alignment algorithm for homology modeling and threading. Protein Sci. 1998 Feb;7(2):254-8
Homology Modeling: Basic Idea: • Similar sequence=> Similar structure • Structure is conserved more than sequence • Structure of new protein derived using existing protein structures as templates. • Changes are compensated for locally.
Homology Modeling: Twilight Zone: below 25% sequence homology
Homology Modeling: • Similar sequence=> Similar structure
Homology Modeling: Step One: • Align sequence of your protein (unknown) with that of candidate template proteins (known)
Homology Modeling: Step Two: • Select template proteins based on sequence similarity and minimize their X-ray structures • The whole sequence can be matched by one or more templates
Homology Modeling: Step Three: • Combine the main chain of the template proteins and fill-in gap sections to generate a complete main chain model of your protein • Gaps are filled-in by using short sequences from a sequence linker library, the selected short
Homology Modeling: Step Three: • Combine the main chain of the template proteins and fill-in gap sections to generate a complete main chain model of your protein • Gaps are filled-in by using short sequences from a sequence linker library, the selected short sequences need to be exchangeable to the section of your original protein.
Homology Modeling: • Step Four: Adding side chains to the main-chain model based on the sequence of your protein: • Mutate and add
Homology Modeling: Step Five: • Minimization and MD of the homology model of your protein
Homology Modeling: Swiss-Model - an automated homology modeling server developed at Glaxo Welcome Experimental Research in Geneva. http://www.expasy.ch/swissmod/ Closely linked to Swiss-PdbViewer, a tool for viewing and manipulating protein structures and models. Likely take 24 hours to get results returned!
Homology Modeling: How Swiss-model works? • 1) Search for suitable templates • 2) Check sequence identity with target • 3) Create ProModII jobs • 4) Generate models with ProModII • 5) Energy minimization with Gromos96 • First approach mode (regular) • First approach mode (with user-defined template) • Optimize mode
Homology Modeling: How Swiss-model works? Program Database Action BLASTP2 ExNRL-3D Find homologous sequences of proteins with known structure. SIM -- Select all templates with sequence identities above 25%. -- -- Generate ProModII input files ProModII ExPDB Generate all models Gromos96 -- Energy minimization of all models
Threading Methods: Similar proteins at the sequence level may have very different secondary structures. On the other hand, proteins very different at the sequence level may have similar structures. Why? Because the protein function is determined by its functional sites, which reside in the cores not the loops. Therefore, researchers propose the inverse protein folding problem, namely, fitting a known structure to a sequence. The problem of aligning a protein sequence to a given structural model is known as protein threading. Given a protein whose structure is known, we derive a structural model by replacing amino acids by place-holders, each is associated with some basic properties such as an alpha-helix or beta-strand or loop of the original amino acids.
Threading Methods: References and software: • Lemer C., Rooman, M. J. & Wodak, S. J. (1996), Protein Structure Prediction By Threading Methods: Evaluation Of Current Techniques, PROTEINS: Structure, Function and Genetics, 23, 337-355. • Bryant, S. H. & Lawrence, C. E. (1993), An empirical energy function for threading a protein sequence through the folding motif, PROTEINS: Structure, Function and Genetics, 16, 92-112. • Alexandrov NN, Luethy R. Alignment algorithm for homology modeling and threading. Protein Sci. 1998 Feb;7(2):254-8 • Jones, D.T., Taylor, W.R & Thornton, J.M (1992), A new approach to protein fold recognition, Nature,358, 86-89. (THREADER).
Threading Methods: Threading methods take the amino acid sequence of an uncharacterized protein structure, rapidly compute models based on a large set of existing 3D structures. The algorithm then evaluates these models to determine how well the unknown amino acid “fits” each template structure. All the threading models in the second to most recent CASP competition produced accurate models in less than half of the cases. However, threading is more successful than homology modeling when attempting to detect remote homologies that can’t be detected by standard sequence alignment.
Threading Methods: Protein Threading Model • Input: • A protein sequence A with n amino acids • A structural model with m core segments Ci: • (1) Each core segment Ci has length ci. • (2) Core segments Ci and Cj are connected by loop Li, which has length between li-min and li-max. • (3) The local structural environment for each amino acid position, such as chemical properties and spatial constraints. • A score function to evaluate a given threading. • Output: • T = {t1, t2, ..., tm} of integers, where ti is the amino acid position in A that occupies the first position in core segment Ci.
Threading Methods: Protein Threading Model • An algorithm: Branch and bound • Spatial constraints: 1 + SUM (cj + lj-min) <= ti <= n + 1 - SUM (cj + lj-min) j < i j >= i ti + ci + li-min <= ti+1 <= ti + ci + li-max • A score function (second order, considering pairwise interaction): f(T) = SUM g1(i,ti) + SUM g2(i,j,ti,tj) i j > i • Algorithm testing: self-threading and using structural analogs.
Ab initio Methods: • ab initio means from the beginning. • Ab-initio algorithms attempt to predict structure based on sequence information alone (i.e., no emperical structural info is considered). • Although many researchers are working in this vein, it is a science in progress – sometimes marginally successful, but very unreliable. • Methods: MD and Simplified models
Ab initio Methods: References: • Hardin C, Pogorelov TV, Luthey-Schulten Z. Ab initio protein structure prediction. Curr Opin Struct Biol. 2002 Apr;12(2):176-81. Review. • Srinivasan R, Rose GD. Ab initio prediction of protein structure using LINUS. Proteins. 2002 Jun 1;47(4):489-95. • Bonneau R, Strauss CE, Rohl CA, Chivian D, Bradley P, Malmstrom L, Robertson T, Baker D. De novo prediction of three-dimensional structures for major protein families. • J Mol Biol. 2002 Sep 6;322(1):65-78. • Bystroff C, Shao Y. Fully automated ab initio protein structure prediction using I-SITES, HMMSTR and ROSETTA. Bioinformatics. 2002 Jul;18 Suppl 1:S54-61
Ab initio Methods: LINUS as an example: Local Independently Nucleated Units of Structure • 50 amino acids are folded at a time, in an overlapping fashion: 1-50, 26-75, ... • Based on the idea that actual proteins fold by forming local secondary structure first. • Side chains are simplified. Only 3 interactions are used: • 1 repulsive: steric • 2 attractive: H-bonds and hydrophobic • Then the calculation of all possibilities for the search of the lowest free energy