Download
1 / 1
10 likes | 84 Vues
Production of polypeptides, 3000-10000 Da, and middle-down analysis by LC-MSMS Catherine Fenselau 1 , Joseph Cannon 1 , Nathan Edwards 2 , Karen Lohnes 1 , Colin Wynne 1

E N D
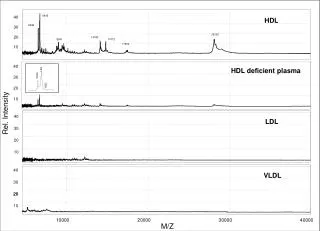
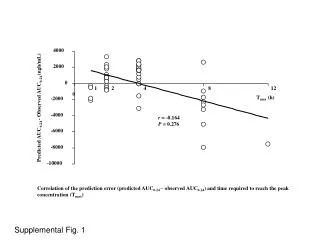
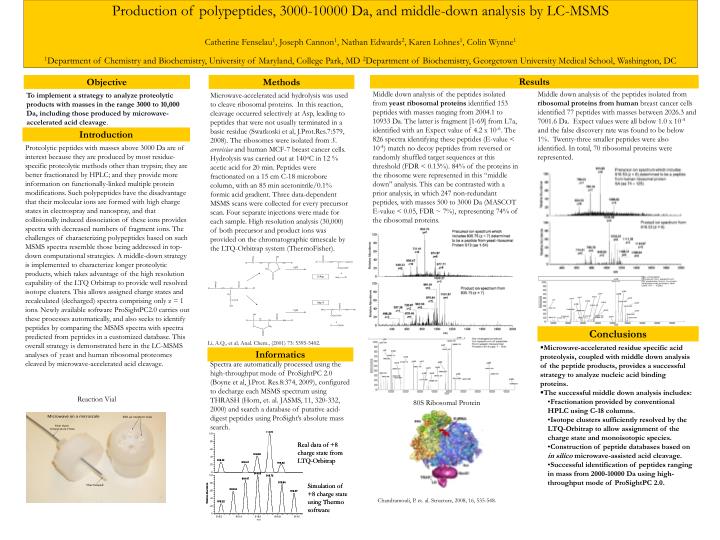
Production of polypeptides, 3000-10000 Da, and middle-down analysis by LC-MSMS Catherine Fenselau1, Joseph Cannon1, Nathan Edwards2, Karen Lohnes1, Colin Wynne1 1Department of Chemistry and Biochemistry, University of Maryland, College Park, MD 2Department of Biochemistry, Georgetown University Medical School, Washington, DC H2O + X-Asp Asp-X H2O + Objective Methods Results Middle down analysis of the peptides isolated from yeast ribosomal proteins identified 153 peptides withmasses ranging from 2004.1 to 10933 Da. The latter is fragment [1-69] from L7a, identified with an Expect value of 4.2 x 10-6. The 826 spectra identifying these peptides (E-value < 10-4) match no decoy peptides from reversed or randomly shuffled target sequences at this threshold (FDR < 0.13%). 84% of the proteins in the ribosome were represented in this “middle down” analysis. This can be contrasted with a prior analysis, in which 247 non-redundant peptides, with masses 500 to 3000 Da (MASCOT E-value < 0.05, FDR ~ 7%), representing 74% of the ribosomal proteins. Middle down analysis of the peptides isolated from ribosomal proteins from human breast cancer cells identified 77 peptides with masses between 2026.3 and 7001.6 Da. Expect values were all below 1.0 x 10-4 and the false discovery rate was found to be below 1%. Twenty-three smaller peptides were also identified. In total, 70 ribosomal proteins were represented. To implement a strategy to analyze proteolytic products with masses in the range 3000 to 10,000 Da, including those produced by microwave-accelerated acid cleavage. Microwave-accelerated acid hydrolysis was used to cleave ribosomal proteins. In this reaction, cleavage occurred selectively at Asp, leading to peptides that were not usually terminated in a basic residue (Swatkoski et al, J.Prot.Res.7:579, 2008). The ribosomes were isolated from S. cerevisiae and human MCF-7 breast cancer cells. Hydrolysis was carried out at 140oC in 12 % acetic acid for 20 min. Peptides were fractionated on a 15 cm C-18 microbore column, with an 85 min acetonitrile/0.1% formic acid gradient. Three data-dependent MSMS scans were collected for every precursor scan. Four separate injections were made for each sample. High resolution analysis (30,000) of both precursor and product ions was provided on the chromatographic timescale by the LTQ-Orbitrap system (ThermoFisher). Introduction Proteolytic peptides with masses above 3000 Da are of interest because they are produced by most residue-specific proteolytic methods other than trypsin; they are better fractionated by HPLC; and they provide more information on functionally-linked multiple protein modifications. Such polypeptides have the disadvantage that their molecular ions are formed with high charge states in electrospray and nanospray, and that collisionally induced dissociation of these ions provides spectra with decreased numbers of fragment ions. The challenges of characterizing polypeptides based on such MSMS spectra resemble those being addressed in top-down computational strategies. A middle-down strategy is implemented to characterize longer proteolytic products, which takes advantage of the high resolution capability of the LTQ Orbitrap to provide well resolved isotope clusters. This allows assigned charge states and recalculated (decharged) spectra comprising only z = 1 ions. Newly available software ProSightPC2.0 carries out these processes automatically, and also seeks to identify peptides by comparing the MSMS spectra with spectra predicted from peptides in a customized database. This overall strategy is demonstrated here in the LC-MSMS analyses of yeast and human ribosomal proteomes cleaved by microwave-accelerated acid cleavage. Conclusions Li, A.Q., et al, Anal. Chem., (2001) 73: 5395-5402. • Microwave-accelerated residue specific acid proteolysis, coupled with middle down analysis of the peptide products, provides a successful strategy to analyze nucleic acid binding proteins. • The successful middle down analysis includes: • Fractionation provided by conventional HPLC using C-18 columns. • Isotope clusters sufficiently resolved by the LTQ-Orbitrap to allow assignment of the charge state and monoisotopic species. • Construction of peptide databases based on in silico microwave-assisted acid cleavage. • Successful identification of peptides ranging in mass from 2000-10000 Da using high-throughput mode of ProSightPC 2.0. Informatics Spectra are automatically processed using the high-throughput mode of ProSightPC 2.0 (Boyne et al, J.Prot. Res.8:374, 2009), configured to decharge each MSMS spectrum using THRASH (Horn, et. al. JASMS, 11, 320-332, 2000) and search a database of putative acid-digest peptides using ProSight’s absolute mass search. Reaction Vial 80S Ribosomal Protein Chandramouli, P. et. al. Structure, 2008, 16, 535-548.