Download

1 / 110
1.16k likes | 2.88k Vues
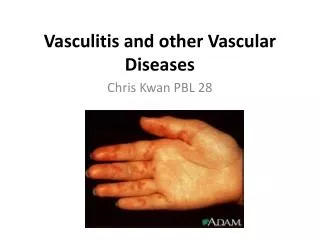
Cutaneous Vascular Diseases. Michael Hohnadel, DO 10/11/05. Cutaneous small-vessel vasculitis (leukocytoclastic vasculitis). Presentation: Palpable purpura ranging in size from a pinpoint to several centimeters Early on lesion may not be palpable

E N D
Cutaneous Vascular Diseases Michael Hohnadel, DO 10/11/05
Cutaneous small-vessel vasculitis(leukocytoclastic vasculitis) • Presentation: Palpable purpura ranging in size from a pinpoint to several centimeters • Early on lesion may not be palpable • Papulonodular, vascular, bullous, pustular or ulcerated forms may develop • Predominate on the ankles and lower legs • dependent areas • Mild pruritis, fever, malaise, arthralgia and/or myalgia may occur. Renal, GI
Cutaneous small-vessel vasculitis(leukocytoclastic vasculitis) • Course: • Typically resolve in 3 to 4 weeks with postinflammatory hyperpigmentation • May recur or become chronic. • Histology: Angiocentric segmental inflammation, endothelial cell swelling, fibrinoid necrosis of blood vessel walls and a cellular infiltrate composed of neutrophils showing fragmentation of nuclei.
Cutaneous small-vessel vasculitis(leukocytoclastic vasculitis) • Etilology • Many forms of small-vessel vasculitis are felt to be caused by circulating immune complexes. These lodge in vessel walls and activate compliment • Infections - bacterial and viral, Drugs, autoimmune disease, lymphoproliferative disorders and solid tumors.
Cutaneous small-vessel vasculitis(leukocytoclastic vasculitis) Workup: • CBC, strep throat culture or ASO titer, Hep B & C serologies and ANA are a reasonable initial screen.
Cutaneous small-vessel vasculitis Treatment • Initial treatment should be nonaggressive • Rest and elevation of the legs • Analgesics, a good diet, and avoidance of trauma or cold • Any identified antigen or drug should be eliminated
Cutaneous small-vessel vasculitis Treatment: • A variety of systemic treatments may be required for severe, intractable or recurrent disease • For disease limited to the skin NSAIDs, antihistamines, colchicine and dapsone. • Systemic corticosteroids for those with systemic manifestations or necrotic lesions • Immunosuppressive agents for rapidly progressive course and severe systemic involvement
LCV • A. classical purpuric papules & papules on lower leg • B. CSVV evolving to form confluent hemorrhagic plaque on posteroir ankle • C. lesions in various stages of evolution
Henoch-Schönlein Purpura(HSP) (anaphylactoid purpura) • Presentation: Characterized by intermittent purpura, arthralgia, abdominal pain, and renal disease • 40 % preceded by mild fever, headache, joint symptoms, and abdominal pain for up to 2 weeks • Typically purpura appears on the extensor surfaces of the extremities • Become hemorrhagic within a day and fades in 5 days • New crops appear over a few weeks
Henoch-Schönlein purpura • Primarily occurs in male children • Peak age 4-8 years • Adults may be affected. • Etiology: Viral infection or streptococcal pharyngitis are the usual triggering event. • May be related to a medications. • Complications: • Pulmonary hemorrhage, Abdominal pain and GI bleeding. • Renal manifestations may occur in 25% or more • Hematuria • Good Prognosis but should be monitored long term for IgA nephropathy
Henoch-Schönlein purpura Treatment : • Supportive • Duration of illness is typically 6 to 16 weeks • 5 and 10 % of patients will have persistent or recurrent disease • Antispasmodics, antibiotics, and antiinflammatory drugs, including systemic corticosteroids • Plamaphoresis in severe cases
Acute Hemorrhagic edema of infancy • Presentation: • Child under the age of 2 with a recent history of an upper respiratory illness (75%) , a course of antibiotics of both. • Abrupt onset of large cockade, annular, or targetoid purpuric lesions involving the face, ears, and extremities • Early in the course there may first be acral edema, may be nontender and asymmetrical • Low-grade fever is common. • No extracutaneous involvement. Children are often nontoxic in appearance.
Acute Hemorrhagic edema of infancy • Etiology: Considered a variant of leukocytoclastic vasculitis with many similarities to HSP • Routine lab tests are nondiagnostic • DDX: meningococcemia, HSP, erythema multiforme, urticaria and Kawasaki’s disease • Clinically most urgent to exclude meningococcemia • Course: Spontaneously resolves with 1-3 weeks
Acute Hemorrhagic edema of infancy • Multiple erythematous, nummular & targetoid plaques on an infant’s thighs
Urticarial vasculitis • Three clinical features distinguish the skin lesions of urticarial vasculitis from urticaria • Lesion are usually painful rather than pruritic • Lesions last longer than 24 hours • On healing there is postinflammatory hyperpigmentation • Systemic symptoms. • Assoc with hypocomplementemia (in some cases), arthritis, angioedemia, abdominal/chest pain.
Urticarial vasculitis • Several erythematous urticarial plaques on the foot & ankle
Urticarial vasculitis • Many disease associations: gammopathies, SLE, viral (hepatitis, EBV). • Treatment: • Pts with hypocomplementemic type respond to oral corticosteroids • Hydroxychloroquine sulfate, colchicine, dapsone, NSAIDs or pentoxifylline • Some pts require a combination of therapies with antihistamines
Familial Mediterranean fever • A periodic fever syndrome reported to affect Sephardic Jews, Armenians, and individuals of Arabian descent. • Presentation: Cutaneous findings consist of erysipelas-like erythema showing a sharp border. • Erysipelas-like erythema occurs in less than half of patients • Affects the lower extremities on the dorsa of the feet , over the ankles, and sometimes the knees • Arthralgias, peritonitis, and constipation may occur • No lymphadenopathy
Familial Mediterranean fever • Onset: Usually under 10 years • Biopsy: leukocytoclastic vasculitis • Tx - colchicine
Erythema elevatum diutinum • A rare condition considered to be a chronic fibrosing leukocytoclastic vasculitis • Presentation: Multiple yellow papules develop over the joints, particularly the elbows, knees, hands, and feet • Initially nodules are soft & mobile then take on a doughy to firm consistency and develop red to purple. • Face & ears usually affected • Lesions may be painful or asymptomatic • Course: Variable, lasting 5-35 yrs. • Assoc: HIV. Gammopathies. • TOC: Dapsone
Top: multiple symmetric red-brown nodules & plaques on extensor surface of digits • Bottom: red-brown palques & nodules of concha, antihelix & of the ear
Erythema elevatum diutinum • Early stage: dense perivascular infiltrate of neutrophils admixed with lymphs & histiocytes • Late-stage: minimal inflammatory infiltrate & marked perivascular fibrous thickening
Granuloma Faciale • Presentation: Brownish-red, infiltrated papules, plaques, and nodules typically on the face esp nose. (head or neck) • Typically healthy, middle aged, white men • Pathology: Identical to EED. Mixed vasculitis with less prominent fibrosis. • Treatment • Often resistant to TX. • Intralesional corticosteroids (first line non-scarring option) • Also: Cryotherapy, dermabrasion, electrosurgery, many others.
Granuloma faciale Note prominent follicular openings
Granuloma faciale • Left: dense, diffuse dermal infiltrate with a Grenz zone • Right: high power of polymorphous infiltrate of lymphocytes, eos, neuts & plasma cells
Polyarteritis Nodosa • Characterized by necrotizing vasculitis affecting the small and medium-sized muscular arteries • Two major forms: • Systemic • Microscopic polyangiitis • Benign cutaneous
Polyarteritis nodosa Systemic PAN • Cutaneous findings (40 %) • 15% of pts have 5 - 10 mm subcutaneous nodules occurring singly or in groups distributed along the course of blood vessels. • Skin above is normal or slightly erythematous • Often painful, may pulsate or ulcerate
PAN • Top: petechiae & multiple purpuric papules with central necrosis on plantar surface • Bottom: confluent hemorrhagic plaques on the medial & plantar aspect of the foot
Polyarteritis nodosa Systemic PAN • Internal manifestations: • May involve the vessels throughout the entire body. • Hypertension, tachycardia, fever, edema glomerulosclerosis and weight loss are the cardinal signs of the disease • Mononeuritis multiplex, most often manifested as foot drop, is the hallmark of PAN
Polyarteritis nodosa Systemic PAN Laboratory findings: • Leukocytosis as high as 40,000 may occur with neutrophilia to 80% • Thrombocytosis and progressive normocytic anemia, elevated sed rate. • Urinary abnormalities seen in 70% • May have elevated C-ANCA (consult Dr. Berrett)
Polyarteritis nodosa Microscopic Polyangiitis • Considered by many to be a subset of systemic PAN. • May be related to Wegner’s and Churg- Strauss. • Segmental, necrotizing and crescentic glomerulonephritis associated with extrarenal vasculitis involving small-size vessels without granulomas or asthma • Positive P-ANCA
Polyarteritis nodosaepidemiology Systemic PAN • 4 X more common in men than women. Mean age 45 yrs. • Assoc with: IV drug abusers, SLE, inflammatory bowel disease, hairy cell leukemia, and familial Mediterranean fever. Also: Hep-B and Hep-C. (check ck for during work-up)
Polyarteritis nodosa Systemic PAN • Histology: An inflammatory necrotizing and obliterative panarteritis that attacks the small and medium-sized arteries. • Biopsy is mainstay of diagnosis.
Polyarteritis nodosa Systemic PAN Course: • Untreated classic PAN has a 5 year survival rate of 13%. • Death from renal failure, cardiovascular or GI complications Treatment: • High-dose corticosteroids with slow taper to DC. • Cytotoxic agents such as may be added like cyclophosphamide with taper to DC.
Polyarteritis nodosa Cutaneous PAN • Absence of visceral involvement • Presentation: Recurrent skin, joint, and muscle involvement without involvement of vital organs • Cutaneous findings similar to those described for the systemic form • Assoc: Hep B,C. Crohn’s & others. • Most patient respond well to aspirin, prednisone, methotrexate, alone or in combination
Wegener’s Granulomatosis • Syndrome consisting of: • Necrotizing granulomas of the upper and lower respiratory tract. • Generalized necrotizing angiitis affecting the medium-sized blood vessels. • Focal necrotizing glomerulitis.
Wegener’s granulomatosis • Presentation: Rhinorrhea, severe sinusitis, and nasal mucosa ulcerations. One or several nodules in the nose, larynx, trachea, or bronchi. • Fever, weight loss and malaise • m:f = 1.3 : 1 Age 40 to 50 years. • “strawberry gums” = hypertrophic gingivitis • Focal necrotizing glomerulitis occurs in 85% of patients • Cutaneous findings (45% of patients) • Nodules may appear in crops, especially along the extensor surface of the extremities • Firm,slightly tender, flesh-colored or violaceous nodules may later ulcerate
Wegener’s granulomatosis • LAB: (+) C – ANCA • Histology: Cutaneous lesions may demonstrate a leukocytoclastic vasculitis with or without granulomatous inflammation.
Wegener’s granulomatosis Nodules on extensor surfaces
Wegener’s granulomatosis • Left: purpuric plaques on the distal fingers • Right: ulceration of the tongue
Wegener’s granulomatosis Prognosis and treatment: • Untreated the mean survival time is 5 months and a 90% mortality over 2 years. • Improves when corticosteroids are combined with cytotoxic drugs: • 75% remission rate, 87% survivalrate in pts between 6 months & 24 yrs
Churg-Strauss Syndrome Presentation: • 3 clinical phases occur: • Prodrome: asthma, allergic rhinitis, peripheral blood eosinoiphilis & eosinophilic infiltrative dx • 2 – 12 years of prodrome. • Vasculitis, characterized by arthritis, & myositis with cardiac, pulmonary, nervous system, GI, renal, ocular or genitourinary dx. • Post-vasculitic phase-dominated by allergic rhinitis, asthma, HTN & peripheral nerve damage
Churg-Strauss Syndrome • Cutaneous lesions • Most common features of the vasculitic phase (70%) • Most common skin findings are purpura & infiltrated nodules on extensor surfaces. • Less common are: necrotizing livedo reticularis, migratory erythema, new onset Raynaud’s phenomenon & digital ischemia. • Extracutaneous manifestations: wt loss, myalgias, arthralgias, mononeuritis multiplwex, GI symptoms, & cardiomyopathy
Churg-Strauss Syndrome • Lab • Peripheral eosinophilia and (+) P-ANCA (less frequently for C-ANCA) • Correlate with disease severity • Histopathology: Sm. vessel vasculitis with eosinophils. • Etiology: Several drugs have been implicated: Zafirlukast, Azithromycin, freebase cocaine. • TX: prednisone alone clinical remission in >90% of pts
Churg-Strauss Syndrome • Crusted, firm papules of the elbow