Download

1 / 30
300 likes | 522 Vues
Importance of early diagnosis of Sickle Cell Anaemia. Dr. J.O. Lawson FWACP, MScPH Consultant Paediatrician Zankli Medical Centre, Abuja. Outline. Introduction Aetiology / Pathophysiology Prevalence of sickle cell disorder in Nigeria Clinical Presentation Benefits of early diagnosis

E N D
Importance of early diagnosis of Sickle Cell Anaemia Dr. J.O. Lawson FWACP, MScPH Consultant Paediatrician Zankli Medical Centre, Abuja
Outline • Introduction • Aetiology /Pathophysiology • Prevalence of sickle cell disorder in Nigeria • Clinical Presentation • Benefits of early diagnosis • Conclusion
Introduction • Sickle Cell Disease is the commonest inherited disorder in the world • It is a single-gene defect that results in sickle-shaped red blood cells that can cause multisystem morbidity and increased risk of early death • It is a relentless killer of people of African, Asian, Caribbean, Mediterranean and Middle Eastern origin • More than 13 million individuals world-wide suffer from sickle cell disease • The prevalence of the sickle-cell trait is between 10% and 30% in West Africa
Introduction • 300,000 infants are born with sickle-cell anaemia in Africa every year, with Nigeria accounting for about half of these • Mortality is high, especially in the early childhood years • 1-2% of the population affected and about 25-30% born with sickle cell trait (35million) • SCA contributes to 5% of underfive deaths on the African continent and up to 16% of under-five deaths in Nigeria • About 60,000 people in the USA and 10,000 in the UK have the disease
Estimates for sickle cell trait prevalence in Nigeria 29% of >5yrs (Fleming 1979) 25-28% (WHO, 1994) 23.7% (Omotade et al, 1998) 17.14% (AKTH, Kano, 2006) • By the share size of Nigeria, it is one of the most affected countries in the world • No reliable database on sickle cell disorders and trait exists in the Ministry of Health • A clearer understanding of the proportionate contribution of SCD to mortality will greatly inform public health Mgt strategies
Major sickle genotypes • HbSS disease or sickle cell anemia - Homozygote for the S globin with usually a severe or moderately severe phenotype with the shortest survival • HbSCdisease - Double heterozygote for HbS and HbC characterized by moderate clinical severity • HbS/B° thalassemia - Double heterozygote for HbS and B° thalassemia and clinically indistinguishable from sickle cell anemia (SCA) • HbS/B+ thalassemia - Mild-to-moderate severity with variability in different ethnicities • HbS/hereditary persistence of fetal Hb (S/HPHP) - Very mild or asymptomatic phenotype • HbS/HbE syndrome - Very rare with a phenotype usually similar to HbS/B+ thalassemia • Rare combinations of HbS with other abnormal hemoglobins such as HbD Los Angeles, G-Philadelphia, HbO Arab, and others
Each parent has two alleles (parts of chromosomes) for hemoglobin and they pass one allele to each child For two adults with sickle cell trait, for each pregnancy there is: 25 percent chance of having a child with sickle cell anaemia(SS) 25 percent chance of having a child with neither sickle cell trait nor SCD(AA) 50 percent risk of having a child with sickle cell trait(AS)
Aetiology of SCA • Blood disorder characterized by the presence of abnormal haemoglobin. • A point mutation of the B-globin gene yielding a substitution of valine for glutamine at the 6th codon and the production of Hemoglobin S • The resulting Hb has the physical properties of forming polymers under reduced oxygen conditions • It also exhibits changes in solubility and molecular stability • These properties are responsible for the profound clinical expressions along with problems of cell volume regulation and endothelial adhesion
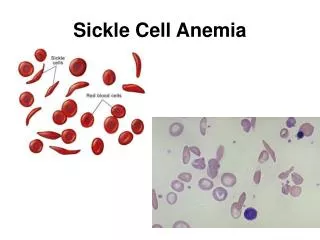
Pathophysiology • RBCs in SCA contain mostly hemoglobin S do not live as long as normal red blood cells (15-50 days) • In the deoxygenated state, the substituted valine on HbS binds to other adjacent β globin molecules, resulting in highly ordered molecular polymers • Cells become stiff, distorted in shape and have difficulty passing through the body’s small blood vessels, blocking small blood vessels with reduced blood supply • With repeated sickling, the rbc membrane eventually becomes permanently damaged and irreversible sickling occurs • These sickle cells become trapped in smaller blood vessels and prevent the flow of blood to various parts of the body
Pathophysiology • The clinical manifestations of SCA are mainly due to repeated vaso-occlusion, chronic intravascular haemolysis, microvascular ischemia and organ damage • One of the most detrimental effects of sickling is vaso-occlusion within the spleen, which results in functional asplenia in 94% of SCA patients by the age of 5 years. In early life, splenomegaly has been known to be common • In later years, however, "autosplenectomy" commonly occurs, the spleen becomes firm, smaller, nodular and finally reduced into a small remnant • With functional asplenia, the patient can no longer filter waste products such as damaged sickle cells or bacteria from the blood. Younger children with SCD are often more susceptible to infection
History of survival • 1960 SCD used to be a ‘disease of children’ • 1973, median survival was 14.3 yrs, 20% died before 2 yrs, one third before 5 yrs, half btw 5-30 yrs • 30yrs ago, only one half of children with SCD were expected to reach adulthood • 1994 Platt et al Median age at death 42 (M) 48(F), 50% survived beyond 50yrs in Boston • CSSCD: 85% of SS and 95% of SC survived 20yrs • Wierenga et al (Jamaica 2001) Median survival calculated was 53 years (95% CI 49.3-57.0) for men and 58.5 (55.1-67.5) for women • Telferet al 2007 estimated survival of children with HbSS in UK study at 16 years was 99.0% • However, no firm data on the survival of patients with SCA on the African continent
Improvement in survival • Quinn et al (2010)showthat the life expectancy of children with sickle cell diseasehas indeed improved • In their careful study of a large cohort,identified by newborn screening and treated in a single comprehensivesickle cell centre, the predicted overallsurvival of patients with Hb SS and Hb SB° at 18 years of age to be 86% • In many developed countries, most children with sickle cell anemia (93.9%) and nearly all children with milder forms of SCD (98.4%) now live to become adults • A large portion of this reduction is attributable to newborn screening programs, the earlyintroduction of prophylactic antibiotics, and the availabilityof effective vaccines for the prevention of life-threateningbacterial infection
Temporal changes in causes of death in children with sickle cell disease. Dallas Newborn Cohort (1983-2007 Analysis)
Sickle Cell Disease in Africa:High childhood mortality Fleming, 1979: Nigeria, < 2% of children with SCA survived beyond 5 years in a rural community Athale and Chintu, 1994: Zambia SCD 2.92% of pediatric admissions; but case fatality 6.61%, (55% <5yr), SCA-related mortality peaked in the 1-5 year old age group (38.71%) followed by the 6-10 year old age group (20.97%) Thuilliez and Vierin, 1997: Gabon 8.4% of under five deaths due to SCD. Koko, et al., 1998: 7.2% of deaths <5 yrs due to SCD SenegalDiagne et al, 2000 Senegal1.1 % mortality per year of follow up Adarangani et al 2009 Kenya 1% mortality per year
Nigeria • Survival data are not available for children with sickle cell disease (SCD) • The few previous childhood SCD studies do not reflect the benefits of modern therapy • Accurate survival data for children with SCD facilitate treatment and counseling of patients and their families, guide public health interventions, and provide the foundation for future research • The best method to measure overall survival is a cohort study of subjects identified at birth or in the first few months of life
Clinical presentation • Usually > 4 months old • Variations in the severity and number of manifestations • The most common signs and symptoms linked to anemia: fatigue, paleness, easy fatigability or shortness of breath • Jaundice • Frequent Infection • Painful crises: may occur in any part of the body (Dactylitis may be the first sign of sickle cell anemia) • Acute chest syndrome: Inflammation, infection, and occlusion of small vessels may cause this syndrome. Signs include chest pain, coughing, difficulty breathing, and fever • Splenic sequestration crises: The spleen becomes enlarged by trapping (or "sequestering") the abnormal RBCs
Clinical presentation • Stroke: Impaired blood flow in the brain can occur when the sickle-shaped cells block small blood vessels, which may lead to a stroke. Signs can include headache, seizures, weakness of the arms and legs, speech problems, a facial droop, or loss of consciousness • Aplastic crisis: bone marrow temporarily slows its production of RBCs due to infection or another cause, resulting in a serious drop in RBCs and severe anemia • Severe or long-lasting anemia can damage the heart, brain, lungs, kidney, spleen, and other organs of the body • Other possible complications include leg ulcers, bone or joint damage, gallstones, kidney damage, painful prolonged erections in males (priapism) leading to erectile dysfunction, eye damage, and delayed growth.
Many patients with sickle cell disease require occasional blood transfusions • Patients with severe complications (such as stroke and acute chest syndrome) may require months or years of regular transfusions to prevent ongoing damage • Hydroxyureamay be helpful in reducing crises and the need for transfusions • Simple ultrasound of the head (TCD)can identify children at high risk for strokes • Regular checkups to detect eye damage necessary
Benefits of early diagnosis • Some complications can be prevented with early diagnosis and treatment. Sickle cell disease and sickle cell trait can be diagnosed with a simple blood test • In the United States testing for sickle cell disease is a part of their newborn screening program • It is possible to diagnose sickle cell disease before birth by amniocentesis or chorionic villus sampling • The goals of treating sickle cell disease are to prevent or relieve pain; prevent infections, organ damage, and strokes; treat anemia; and control complications • Counseling, prophylaxis, immunizations, follow up an information on new developments
Newborn Screening for Sickle Cell Diseasein Africa WHO: • Has recognized SCD as a major public health problem • Has recommended member nations to develop comprehensive programs to “control” SCD, including early diagnosis • No country in Africa has implemented national newborn screening program for sickle cell disease. • Most children with SCD in Africa are not diagnosed before clinical presentation, complications, or death. ALL African and other nations where SCD is common should embark on newborn screening for SCD as a public health program
Newborn Screening for Sickle Cell DiseaseJustification • Relatively common, serious condition with clinical manifestations starting in infancy • SCD most common condition diagnosed in newborn screening in U.S.; age 6 - 12mo had highest mortality rate. • Simple, accurate diagnostic tests available • Isoelectric focusing, HPLC methods highly accurate and simple • Proven life-saving, inexpensive preventive treatment available • Penicillin prophylaxis is simple, inexpensive, and effective. Good evidence that newborn screening followed by comprehensive medical care including parental health education, penicillin prophylaxis, and anti-pneumococcal vaccination have reduced early mortality in SCD
Managing SCD • Go for regular checkup • Comprehensive Immunization • Learn as much as possible about the disease • Good hydration • Protect from extreme heat and cold • Daily Folic acid and anti-malarial • Penicillin prophylaxis in young children • Proper nutrition • Psychological support
Seek medical attention when: • Fever of >38°C • Pain that isn't relieved by oral medication • Chest pain, shortness of breath or difficulty in breathing • Extreme tiredness or somnolence • Severe headaches or dizziness • Severe stomach pain or swelling • Jaundice or extreme paleness • Sudden change in vision • Convulsions • Weakness or inability to move any part of the body • Slurred speech • Loss of consciousness • Numbness or tingling • Painful erection in males
Recent developments • Bone marrow transplants can cure sickle cell disease. Because the procedure has significant risks, transplants are not appropriate for every patient • Bone marrow transplants are used primarily in young patients who have severe sickle cell disease. However, the decision to give this treatment is made on a case-by-case basis • Bone marrow used for a transplant must come from a closely matched donor. This is usually a close family member who doesn't have sickle cell disease • Researchers continue to look for ways to reduce the risks of this procedure and to widen its application
Recent developments • Gene Therapy: • How a normal gene can be put in the bone marrow of a person who has sickle cell disease. This would cause the body to make normal red blood cells. • Researchers also are studying whether they can "turn off" the sickle cell gene or "turn on" a gene that makes red blood cells behave more normally • New Medicines • Some of these new interfere with sickling of hemoglobin, others prevent the cells from sticking to blood vessel walls, and some raise levels of fetal hemoglobin
Bone marrow transplant: the only known cure for sickle cell disease. Transplants are complex and risky procedures and currently are an option only for a carefully selected subset of patients with severe complications • To be eligible, a child would need bone marrow or stem cells from a "matched" donor with a low risk of rejection. Even then, the procedure has significant risks and there is the chance of rejection of the transplanted marrow
Conclusion • SCD continues to be a killer of Nigerians • Advances which are mainly applicable in high resource countries, have unfortunately widened the gap in terms of quality of life between patients in developed countries and those in developing countries • Need to improve the capability of laboratories to make correct diagnosis • Need for education, counseling and support • The long-term follow-up of individuals with SCD is important and Newborn Cohort studies would be a powerful tools to monitor survival into early adulthood and help identify predictors of outcome • It is expected that with early identification by newborn screening, initiation of penicillin and routine immunizations, education of those involved in the child’s care, and lifelong, specialized follow up care, many of the complications of sickle cell disease can be prevented or managed promptly so that individuals can lead healthy, productive lives
Tiffany McCoy • “Having a good attitude affects any area of your life. I always try my best to have a good outlook on life. I have sickle cell disease, but sickle cell doesn't have me”