Download

1 / 43
460 likes | 919 Vues
Energia libera di Gibbs. Sistema isolato:. In generale:. È più comodo dover considerare solo il sistema. Prendiamo un sistema a pressione e temperatura costante. La funzione H-TS deve tendere ad un minimo per trasformazioni spontanee a pressione e temperatura costanti.

E N D
Energia libera di Gibbs Sistema isolato: In generale: È più comodo dover considerare solo il sistema. Prendiamo un sistema a pressione e temperatura costante La funzione H-TS deve tendere ad un minimo per trasformazioni spontanee a pressione e temperatura costanti.
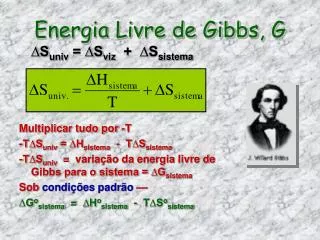
Energia libera di Gibbs G = H – TS A T e p costante: dG = dH-TdS ≤ 0 Per una trasformazione finita (integrazione): G = H - TS All’equilibrio (condizioni di reversibilità): dG = 0 Per una trasformazione spontanea (irreversibilità): dG < 0 Finchè un processo non ha raggiunto l’equilibrio dG < 0 e quindi procede spontaneamente e irreversibilmente verso l’equilibrio fino a quando dG = 0.
Ma attenzione per una trasformazione tra gli stessi stati iniziali e finali: G(rev) = G (irr) L’energia libera è una nuova funzione di stato! G = G(stato finale) – G(stato iniziale) Per una trasformazione ciclica: L’utilità della funzione di Gibbs nasce dal fatto che la gran parte delle reazioni chimiche viene svolta in condizioni di temperatura e pressione costanti.
Per trasformazioni finite: G = H - TS G > 0 processo sfavorito G<0 processo favorito • H<0 S>0 : un processo favorito sia dal punto di vista entalpico, che entropico. → G<0 a tutte le temperature. • H>0 S>0 : un processo guidato dal contributo entropico. →G<0 solo se TS> H • H<0 S<0 : un processo guidato dal contributo entalpico. →G<0 solo se H> TS • H>0 S<0 : un processo sfavorito sia dal punto di vista entalpico, che entropico. →G>0 a tutte le temperature.
L’energia libera è una funzione della pressione e della temperatura G(p,T) In particolare si può dimostrare che: dG = Vdp - SdT A p costante: dG = -SdT A T costante: dG = Vdp Queste equazioni ci daranno modo di studiare la dipendenza dell’energia libera dalla temperatura e dalla pressione!
Il potenziale chimico La condizione di spontaneità per un processo a P e T costante è Sembrerebbe che a P e T costante sia sempre dG=0! In realtà G è una grandezza estensiva e dipende anche dal numero di moli di ciascun componente.
È proprio nei casi in cui varia la composizione del sistema, a P e T costante (reazioni chimiche, transizioni di fase), che la condizione dG<0 è utile per determinare la direzione di un processo. A P e T costante i termini in dP e dT scompaiono: La derivata parziale di G rispetto al numero di moli di un componente si dice potenziale chimico:
Il potenziale chimico A P e T costante: PROCESSO SPONTANEO EQUILIBRIO
Il potenziale chimico Potenziale chimico per un componente puro: Il potenziale chimico del componente puro coincide con la sua energia libera molare.
Dipendenza del potenziale chimico dalla pressione: I volumi molari sono grandezze necessariamente positive, quindi il potenziale chimico aumenta sempre all’aumentare della pressione. Dipendenza del potenziale chimico dalla temperatura: Le entropie molari molari sono grandezze necessariamente positive, quindi il potenziale chimico diminuisce sempre all’aumentare della temperatura.
Potenziale chimico ed equilibrio Equilibrio tra fasi per una sostanza pura Fase = una porzione di sostanza uniforme per composizione chimica e stato di aggregazione. A p e T costanti e in presenza di due fasi e β : All’equilibrio: dG = 0
Poiché: E quindi: All’equilibrio, i potenziali chimici di ogni componente nelle diverse fasi devono essere uguali.
Dipendenza della stabilità di fase dalla temperatura Per un componente: Generalmente: Gm p costante -Sm(solido) -Sm(liquido) solido -Sm(gas) liquido gas Tf Tb T Nei punti di transizione di fase (intersezione delle rette): Fusione: Ebollizione:
Dipendenza della stabilità di fase dalla pressione Per un componente: Generalmente: T costante Gm Vm(gas) Vm(solido) Vm(liquido) solido liquido gas pcond psol p
p liquido solido vapore T Diagrammi di stato (p,T)
Vm(solido)>Vm(liquido) P(atm) liquido Punto critico Punto triplo solido vapore 273.16 T(K) Diagramma di stato dell'acqua Punto triplo = condizioni uniche di pressione e temperatura alle quali coesistono la fase liquida, solida e gassosa. Punto critico = condizioni di temperatura e pressione al di sopra delle quali esiste solo la fase vapore.
Potenziale chimico e reazioni chimiche Per una generica reazione (p, T costanti): 2A + 3B C + 2D La variazione del numero di moli di ciascuna specie non è arbitraria, ma è fissata dalla stechiometria della reazione:
La reazione procede spontaneamente nella direzione dG<0 La reazione procede spontaneamente verso i reagenti La reazione procede spontaneamente verso i prodotti
La reazione è all’equilibrio All’equilibrio la somma dei potenziali chimici dei reagenti deve essere uguale alla somma dei potenziali chimici dei prodotti (ciascuno moltiplicato per il proprio coefficiente stechiometrico).
L'equilibrio chimico Per una generica reazione: 2A + 3B C + 2D All’equilibrio:
Se: La reazione procede verso i prodotti (dnC>0). Se: La reazione procede verso i reagenti (dnC<0). All’equilibrio: dG=0 dG<0 dG<0 dnC> 0 dnC< 0 dG=0
Potenziale chimico di un gas puro Stato standard: stato della sostanza pura alla pressione di 1 bar e alla temperatura T (p0=1).
Potenziale chimico di una miscela di gas Consideriamo un gas ad una certa temperatura T e pressione totale p Se è puro: pi=p Nel caso di una miscela di gas ideali: pi=xip
Dipendenza del potenziale chimico dalla composizione Gas perfetto Soluzioni ideali Le soluzioni reali si avvicinano a questo comportamento quando sono diluite Analogamente a quanto fatto con la fugacità per i gas reali, per le soluzioni ideali si definisce una grandezza detta attività, in modo da mantenere la stessa forma funzionale per il potenziale chimico. Soluzioni reali Dove ai è l’ATTIVITA’ del componente i:
Definiamo l’energia libera standard di reazione: Reagenti: ni<0; prodotti ni>0 Nell’esempio: E la costante di equilibrio: Reagenti: ni<0; prodotti ni>0 Nell’esempio:
Reagenti: ni<0; prodotti ni>0 All’equilibrio: 2A + 3B C + 2D Esplicitando il potenziale chimico:
All’equilibrio: K = costante di equilibrio termodinamica Una grandezza adimensionale, come le attività! L’equilibrio è spostato verso i prodotti L’equilibrio è spostato verso i reagenti Prodotti e reagenti hanno la stessa concentrazione
Energia libera standard Condizioni standard: specie pura, p=1 bar, T fissata. In queste condizioni si parla di energia libera standard, G°. Per convenzione: G°(298K)=0 per tuttigli elementi nel loro stato stabile in quelle condizioni di temperatura e pressione (T=298K, p=1 bar). Energia libera standard di reazione:
Energia libera standard di formazione = variazione di energia libera relativa alla reazione di formazione di un composto a partire dai suoi elementi nel loro stato di riferimento. Esempio: 6 C(s) + 3H2(g) C6H6(l) L’energia libera standard di reazione può essere ottenuta dalla somma delle energie libere di formazione dei reagenti e dei prodotti, ognuna moltiplicata per il coefficiente stechiometrico con cui quella specie compare nella equazione di reazione (positivo per i prodotti, negativo per i reagenti).
Prodotti: j>0 Reagenti: j<0 Esempio: CO(g)+½O2(g)CO2(g) T=298K
Esempio: Solubilizzazione di NaCl in acqua (p=1atm, T=298K) NaCl(s)+ nH2O Na+(aq) + Cl-(aq) H°= 1000 calmol-1 (>0: sfavorisce il processo) S°=10.3 calmol-1K-1 = 10.3 u.e. (>0: favorisce il processo) G° = 1000 - 29810.3 = - 2069 calmol-1 < 0 NaCl si solubilizza in acqua esclusivamente per effetti entropici!
Esempio: Racemizzazione in H2O della L-Alanina (T=298K, p=1atm) L-Ala D-Ala In queste condizioni il processo di racemizzazione è favorito!
Esempio: Sintesi di un dipeptide (T=298K, p=1 bar) Leu + Gly Leu-Gly + H2O -153.16 -126.66 -207.10 -68.32 49.5 26.1 67.2 -16.72 In queste condizioni la reazione è spostata verso i reagenti.
Transizioni di fase Esempio - Fusione dell’ H2O (p =1 atm, T=273.16K) H2O(s)H2O(l) Le transizioni di fase sono processi che avvengono in condizioni di reversibilità, finchè sono presenti entrambe le fasi.
Per un gas reale al posto della pressione si introduce la fugacità, f: f = p Tutti i gas reali hanno lo stesso stato standard di riferimento.
Energia libera e lavoro utile G = H - TS dG = dH – d(TS) = dH – TdS - SdT H = U + pV dH = dU + pdV + Vdp dG = dU + pdV + Vdp – TdS - SdT A T e p costanti: dG = dU + pdV– TdS Per una trasformazione reversibile: dU = qrev +wrev= TdS + wrev dG = TdS + wrev+ pdV– TdS = wrev + pdV
wrev= wmax dG = wmax - w(meccanico) = w(utile) Lavoro utile = lavoro realmente utilizzabile in una trasformazione. Es. lavoro elettrochimico. G = H - TS = wutile G = (calore totale) – (calore entropico) = wutile II principio: mentre è possibile trasformare interamente energia meccanica in energia termica, non è possibile trasformare interamente calore in lavoro.
Dipendenza della energia libera dalla temperatura Esempio – Denaturazione della ribonucleasi. N(forma nativa) D(forma denaturata) S(D)>S(N) → S >0 la reazione è favorita da un aumento di temperatura T=303K: H°=57.2 kcalmol-1 S°= 186 calmol-1K-1 G°=H°-TS°= 57200-(303186) = 57200-56399 = 0.9 kcalmol-1 T=313K: H°=76.0 kcalmol-1 S°= 260 calmol-1K-1 G°=H°-TS°= 76000-(313260) = 76000-81380 = -5.4 kcalmol-1
Esempio – Combustione del glucosio H°(298)=-2818 kJmol-1 G°(298)= - 2862 kJmol-1
Esempio - N2(g) + 3H2(g) 2NH3(g) Il numero di specie allo stato gassoso è minore nei prodotti che nei reagenti: n(gas) = -2 S < 0 V < 0 La reazione sarà favorita da una diminuzione di temperatura o da un aumento di pressione.
Equazione di Gibbs-Helmholtz Alla temperatura T1: G(T1) = H(T1) – T1S(T1) Alla temperatura T2: G(T2) = H(T2) – T2S(T2) Considerando l’entalpia e l’entropia indipendenti dalla temperatura:
Dipendenza dell'energia libera dalla pressione Gas ideale: Fasi condensate: