Download

1 / 9
90 likes | 221 Vues
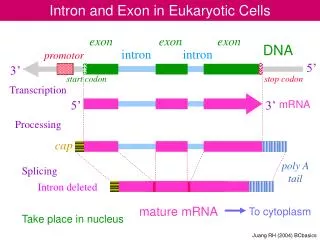
How to tell an exon from an intron?. Overview. Using splicing enhancer WMMs Using repeat statistics Parameters and training sets Structural information. Using Splicing Enhancer (SE) WMMs. 4 WMMs derived from SELEX experiments (M. Zhang & A. Krainer labs, CSHL)

E N D
Overview • Using splicing enhancer WMMs • Using repeat statistics • Parameters and training sets • Structural information
Using Splicing Enhancer (SE) WMMs • 4 WMMs derived from SELEX experiments (M. Zhang & A. Krainer labs, CSHL) • 6051 Refseq genes (“clean” Refseq set) • Scan introns and exons for high-scoring putative SEs. • Correlations of SE positions • Significant differences between exons and introns
Using repeat statistics • ~ 90% of simple and low complexity (LC) repeats in introns are: • AT-rich • [T(A)m]n, [G(A)m]n, [C(A)m]n • Simple and LC repeats in exons are GC-rich
Future Developments • Using SE WMMs with selective repeat masking • An even-periodic, 5th order Markov Model to score introns? • A new Twinscan parameter set based on “cleaner” training set
Parameters and training sets • 3 “clean” Refseq sets (Randy & Sam, Jeltje, Mikhail) • Different selection criteria • Different sizes: 2 sets of ~12000 genes, 1 set of ~6000 genes • Create one training set from these 3 and re-estimate Twinscan parameters