Cystic Fibrosis
Cystic Fibrosis. Paolo Aquino Internal Medicine-Pediatrics January 13, 2005. Outline. What is cystic fibrosis (CF)? What causes CF? What are the manifestations? How do you diagnose CF? How do you treat CF?. Cystic Fibrosis.


Cystic Fibrosis
E N D
Presentation Transcript
Cystic Fibrosis Paolo Aquino Internal Medicine-Pediatrics January 13, 2005
Outline • What is cystic fibrosis (CF)? • What causes CF? • What are the manifestations? • How do you diagnose CF? • How do you treat CF?
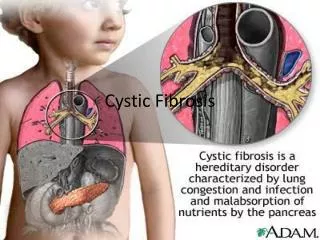
Cystic Fibrosis • Inherited monogenic disorder presenting as a multisystem disease. • Typically presents in childhood • 7% of CF patients diagnosed as adults • Most common life limiting recessive trait among whites
Cystic Fibrosis • Prognosis improving • >38% of CF patients are older than 18 • 13% of CF patients are older than 30 • Median survival • Males: 32 years • Females: 29 years
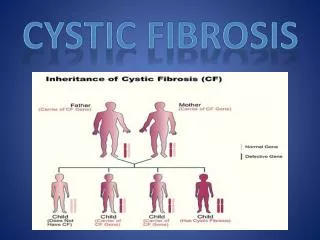
Genetics of CF • Autosomal recessive • Gene located on chromosome 7 • Prevalence- varies with ethnic origin • 1 in 3000 live births in Caucasians in North America and Northern Europe • 1 in 17,000 live births of African Americans • 1 in 90,000 live births in Hawaiian Asians
Genetics of CF • Most common mutation • Occurs in 70% of CF chromosomes • 3 base pair deletion leading to absence of phenylalanine at position 508 (DF508) of the CF transmembrane conductance regulator (CFTR) • Large number (>1000) of relatively uncommon muations (~2%)
Genetics of CF • Difficult to use DNA diagnosis to screen for heterozygotes • No simple physiologic measurements yet available for heterozygote detection
Genetics of CF • The CFTR protein • Single polypeptide chain, 1480 amino acids • Cyclic AMP regulated chloride channel • Regulator of other ion channels • Found in the plasma membrane of normal epithelial cells
Genetics of CF • DF508 mutation leads to improper processing and intracellular degradation of the CFTR protein • Other mutations in the CF gene produce fully processed CFTR proteins that are either non-functional or partially functional
Genetics of CF • Epithelial dysfunction • Epithelia containing CFTR protein exhibit array of normal functions • Volume absorbing (airway, distal intestine) • Salt absorbing without volume (sweat ducts) • Volume secretory (proximal intestine, pancreas) • Dysfunction in CFTR gene leads to different effects on patterns of electrolyte and water transport
Persistence of CF • Is there a reason why CF mutations are so prevalent? • Hypothetical resistance to morbidity and mortality associated with cholera • Evidence shows intestinal epithelial cells homozygous for the DF508 mutation are unresponsive to the secretory effects of cholera toxin
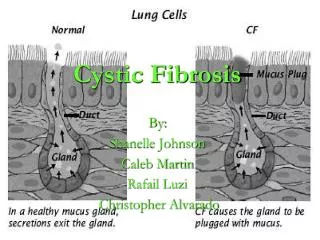
Pathophysiology • Lung • Raised trans-epithelial electric potential difference • Absence of cAMP-dependent kinase and PKC-regulated chloride transport • Raised sodium transport and decreased chloride transport • Alternative calcium-regulated chloride channel in airway epithelia which is a potential therapeutic target
Normal airway epithelia • CF altered airway epithelia
Pathophysiology • Lung • High rate of sodium absorption and low rate of chloride secretion reduces salt and water content in mucus, depletes peri-ciliary liquid • Mucus adheres to airway surface, leads to decreased mucus clearing • Predisposition to Staph and Pseudomonas infections
Pathophysiology • Gastrointestinal • Pancreas • Absence of CFTR limits function of chloride-bicarbonate exchanger to secrete bicarbonate • Leads to retention of enzymes in the pancreas, destruction of pancreatic tissue. • Intestine • Decrease in water secretion leads to thickened mucus and dessicated intraluminal contents • Obstruction of small and large intestines
Pathophysiology • Gastrointestinal • Biliary tree • Retention of biliary secretion • Focal biliary cirrhosis • Bile duct proliferation • Chronic cholecystitis, cholelithiasis • Sweat • Normal volume of sweat • Inability to reabsorb NaCl from sweat as it passes through sweat duct
Manifestations • Common presentations • Chronic cough • Recurrent pulmonary infiltrates • Failure to thrive • Meconium ileus
Manifestations • Respiratory tract • Chronic sinusitis • Nasal obstruction • Rhinorrhea • Nasal polyps in 25%; often requires surgery • Chronic cough • Persistent • Viscous, purulent, green sputum
Manifestations • Respiratory tract • Chronic cough • Exacerbations require aggressive therapy • Postural drainage • Antibiotics • Become more frequent with age • Progressive loss of lung function • Infection • Intially with H. influenzae and S. aureus • Subsequently P. aeruginosa • Occassionally, Xanthomonas xylosoxidans, Burkholderia gladioli, Proteus, E. coli, Klebsiella
Manifestations • Respiratory tract • Lung function • Small airway disease is first functional lung abnormality • Progresses to reversible as well as irreversible changes in FEV1 • Chest x-ray may show hyperinflation, mucus impaction, bronchial cuffing, bronchiectasis
Manifestations • Respiratory tract • Complications • Pneumothorax ~10% of CF patients • Hemoptysis • Digital clubbing • Cor pulmonale • Respiratory failure
Manifestations • Gastrointestinal • Meconium ileus • Abdominal distention • Failure to pass stool • Emesis • Abdominal flat plate • Air-fluid levels • Granular appearancemeconium • Small colon
Manifestations • Gastrointestinal • Meconium ileus equivalent or distal intestinal obstruction syndrome • RLQ pain • Loss of appetite • Emesis • Palpable mass • May be confused with appendicitis
Manifestations • Gastrointestinal • Exocrine pancreatic insufficiency • Found in >90% of CF patients • Protein and fat malabsorption • Frequent bulky, foul-smelling stools • Vitamin A, D, E, K malabsorption • Sparing of pancreatic beta cells • Beta cell function decreases with age • Increased incidence of GI malignancy
Manifestations • Genitourinary • Late onset puberty • Due to chronic lung disease and inadequate nutrition • >95% of male patients with CF have azospermia due to obliteration of the vas deferens • 20% of female patients with CF are infertile • >90% of completed pregnancies produce viable infants
Diagnosis • DNA analysis not useful due to large variety of CF mutations • Sweat chloride test >70 mEq/L • 1-2% of patients with clinical manifestations of CF have a normal sweat chloride test • Nasal transepithelial potential difference
Diagnosis • Criteria • One of the following • Presence of typical clinical features • History of CF in a sibling • Positive newborn screening test • Plus laboratory evidence for CFTR dysfunction • Two elevated sweat chloride concentrations on two separate days • Identification of two CF mutations • Abnormal nasal potential difference measurement
Treatment • Major objectives • Promote clearance of secretions • Control lung infection • Provide adequate nutrition • Prevent intestinal obstruction • Investigation into therapies to restore the processing of misfolded CFTR protein
Treatment • Lung • >95% of CF patients die from complications of lung infection • Breathing exercises • Flutter valves • Chest percussion • ? Hypertonic saline aerosols
Treatment • Lung • Antibiotics • Early intervention, long course, high dose • Staphylococcus- Penicillin or cephalosporin • Oral cipro for pseudomonas • Rapid emergence of resistance • Intermittent treatment (2-3 weeks), not chronic • IV antibiotics for severe infections or infections resistant to orals
Treatment • Lung • Antibiotics • Pseudomonas treated with two drugs with different mechanisms to prevent resistance • e.g. cephalosporin + aminoglycoside • Use of aerosolized antibiotics • Increasing mucus clearance • N-acetylcysteine not clinically helpful • Long-term DNAse treatment increases time between pulmonary exacerbations
Treatment • Lung • Inhaled b-adrenergic agonists to control airway constriction • No evidence of long-term benefit • Oral glucocoticoids for allergic bronchopulmonary aspergillosis • Studying benefits of high dose NSAID therapy for chronic inflammatory changes
Treatment • Lung • Atelectasis • Chest PT + antibiotics • Respiratory failure and cor pulmonale • Vigorous medical management • Oxygen supplementation • Only effective treatment for respiratory failure is lung transplantation • 2 year survival >60% with lung transplatation
Treatment • Gastrointestinal • Pancreatic enzyme replacement • Replacement of fat-soluble vitamins- especially vitamin E & K • Insulin for hyperglycemia • Intestinal obstruction • Pancreatic enzymes + osmotically active agents • Distal- hypertonic radiocontrast material via enema
Treatment • Gastrointestinal • End-stage liver disease- transplantation • 2 year survival rate >50% • Hepatic and gallbladder complications treated as in patient without CF
Summary • CF is an inherited monogenic disorder presenting as a multisystem disease • Pathophysiology is related to abnormal ion transportation across epithelia • Respiratory, GI and GU manifestations • Treatment is currently preventative and supportive