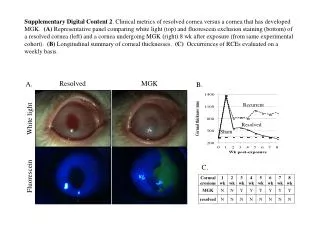
A Fully Resolved Consensus Between Fully Resolved Phylogenetic Trees
A Fully Resolved Consensus Between Fully Resolved Phylogenetic Trees. Jos é Augusto Amgarten Quitzau João Meidanis Scylla Bioinformatics, Brazil University of Campinas, Brazil. Phylogeny reconstruction methods.


A Fully Resolved Consensus Between Fully Resolved Phylogenetic Trees
E N D
Presentation Transcript
A Fully Resolved Consensus Between Fully Resolved Phylogenetic Trees José Augusto Amgarten Quitzau João Meidanis Scylla Bioinformatics, Brazil University of Campinas, Brazil
Phylogeny reconstruction methods • Phylogeny reconstruction methods aim at inferring the phylogenetic tree that best describes the evolutionary history for a set of taxa.
Which tree to choose? • “The field of systematics has been in considerable turmoil as various investigators developed different methods of classification and argued their merits. I guarantee you that no one method or view has all the good points.” Walter M. Fitch – 1984
Consensus as tree constructor • Consensus trees have been used traditionally in tree comparison and calculation of bootstrap values • We propose the use of consensus as a tree constructor • It can be efficiently implemented as long as we keep trees fully resolved
Splits • Every edge in a phylogenetic tree divides the leaves in two subgroups. • Each of these pairs of subgroups are splits of the tree. A B H G C D F E
Tree weight • Our method relies on weighing trees and taking the one with maximum weight • Let the frequency of a split in a collection of trees be the number of trees which contain the split divided by the total number of trees in the collection • Let the weight of an unrooted phylogenetic tree be the product of its splits frequencies
Most probable tree • A most probable tree for a collection of fully resolved phylogenetic trees is a tree that maximizes the weight:
Solution w = 0.0703125
Running time • The tree weight formula can be written as a product of the frequencies of the small subgroups • We designed an algorithm that finds all most probable trees for a given set of fully resolved phylogenetic trees • The complexity of the algorithm is O(l3t2log(lt)),where l is the number of leaves and t is the number of trees
Experiments • Data sets used to test the new method: • Synthetic data: from Gascuel’s LIRMM site • K2P – Kimura 2 Parameter, no MC • K2Pm – Kimura 2 Parameter, with MC • COV – Covarion model, no MC • COVm – Covarion model, with MC • Real data: Ribosomal RNA
Experiments • Programs used to test the new method (19):
Results: average split distance • Consensus consistently yields minimum average split distance
Results: distance to “real” tree • Consensus consistently not worse off than majority of input trees … of input trees
Theoretical foundations A B H G C D F E
All splits of a tree A B A | BCDEFGH H B | ACDEFGH H | ABCDEFG AB | CDEFGH ABCD | EFGH G EFG | ABCDH CD | ABEFGH G | ABCDEFH C | ABDEFGH EF | ABCDGH D | ABCEFGH C F | ABCDEGH D E | ABCDFGH F E
Small subgroup of each split A B A H | BCDEFGH B | ACDEFGH H AB | ABCDEFG | CDEFGH ABCD | EFGH G EFG CD | ABCDH | ABEFGH G C | ABCDEFH | ABDEFGH EF D | ABCDGH C | ABCEFGH F | ABCDEGH D E | ABCDFGH F E
Small subgroups A B H AB ABCD EFG CD G C EF D F E
Maximal clusters (n-trees) A B H AB ABCD EFG CD G C EF D F E
Fundamental theoretical result • The small subgroup set of a phylogenetic tree is always a finite set of n-trees • There are exactly three n-trees in this set, and all n-trees are maximal if and only if the phylogenetic tree is fully resolved ABCD AB CD H A B C D EFG EF G E F
E F G EF GH D ABC Implementation details
a E F G EF GH D ABC Dynamic programming
a b E F G EF GH D ABC Dynamic programming
a b E F G EF GH D ABC Dynamic programming
b a D E E F G DE EF GH D ABC ABC FGH DEF Implementation details L \ba
To Do List • Rooted trees • Polytomies • Non uniform weights for input trees
Acknowledgments • Scylla Bioinformatics and Institute of Computing, Unicamp, for machine time, infrastructure, and support • Brazilian Research Financing Agency CNPq, grant 470420/2004-9