Evolution at the DNA Level: Insights into Mutations, Orthology, and Synteny Mapping
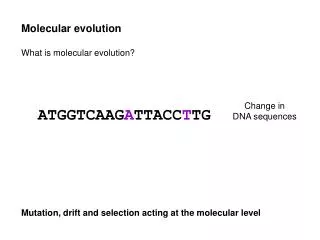
This document explores the evolutionary transformations at the DNA level, focusing on deletion mutations, sequence edits, and rearrangements such as inversions and translocations. It delves into orthologs, paralogs, and the methodologies for constructing synteny maps in various organisms, including humans and mice. Local alignment strategies using tools like BLASTZ, WU-BLAST, and BLAT are highlighted for their effectiveness in genomic comparisons. The statistical approaches for detecting conserved elements through methods like the binomial approach and GERP are discussed, providing a comprehensive overview of DNA evolution and analysis.

Evolution at the DNA Level: Insights into Mutations, Orthology, and Synteny Mapping
E N D
Presentation Transcript
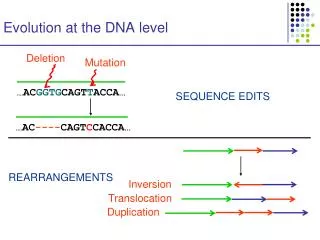
Evolution at the DNA level Deletion Mutation …ACGGTGCAGTTACCA… SEQUENCE EDITS …AC----CAGTCCACCA… REARRANGEMENTS Inversion Translocation Duplication
Orthology and Paralogy Yeast Orthologs:Derived by speciation Paralogs: Everything else HA1 Human HA2 Human WA Worm HB Human WB Worm
Synteny maps Comparison of human and mouse
Building synteny maps Recommended local aligners • BLASTZ • Most accurate, especially for genes • Chains local alignments • WU-BLAST • Good tradeoff of efficiency/sensitivity • Best command-line options • BLAT • Fast, less sensitive • Good for • comparing very similar sequences • finding rough homology map
Index-based local alignment …… Dictionary: All words of length k (~10) Alignment initiated between words of alignment score T (typically T = k) Alignment: Ungapped extensions until score below statistical threshold Output: All local alignments with score > statistical threshold query …… scan DB query Question: Using an idea from overlap detection, better way to find all local alignments between two genomes?
Chaining local alignments • Find local alignments • Chain -O(NlogN) L.I.S. • Restricted DP
Progressive Alignment x • When evolutionary tree is known: • Align closest first, in the order of the tree • In each step, align two sequences x, y, or profiles px, py, to generate a new alignment with associated profile presult Weighted version: • Tree edges have weights, proportional to the divergence in that edge • New profile is a weighted average of two old profiles y Example Profile: (A, C, G, T, -) px = (0.8, 0.2, 0, 0, 0) py = (0.6, 0, 0, 0, 0.4) s(px, py) = 0.8*0.6*s(A, A) + 0.2*0.6*s(C, A) + 0.8*0.4*s(A, -) + 0.2*0.4*s(C, -) Result:pxy= (0.7, 0.1, 0, 0, 0.2) s(px, -) = 0.8*1.0*s(A, -) + 0.2*1.0*s(C, -) Result:px-= (0.4, 0.1, 0, 0, 0.5) z w
Threaded Blockset Aligner HMR – CD Restricted Area Profile Alignment Human–Cow
Reconstructing the Ancestral Mammalian Genome Human: C C Baboon: C G Dog: G C or G Cat: C
Finding Conserved Elements (1) • Binomial method • 25-bp window in the human genome • Binomial distribution of k matches in N bases given the neutral probability of substitution
Finding Conserved Elements (2) A C • Parsimony Method • Count minimum # of mutations explaining each column • Assign a probability to this parsimony score given neutral model • Multiply probabilities across 25-bp window of human genome A A G
Phylo HMMs HMM Phylogenetic Tree Model Phylo HMM
Statistical Power to Detect Constraint N L C: cutoff # mutations D: neutral mutation rate : constraint mutation rate relative to neutral
Statistical Power to Detect Constraint N L C: cutoff # mutations D: neutral mutation rate : constraint mutation rate relative to neutral