Download

1 / 28
280 likes | 468 Vues
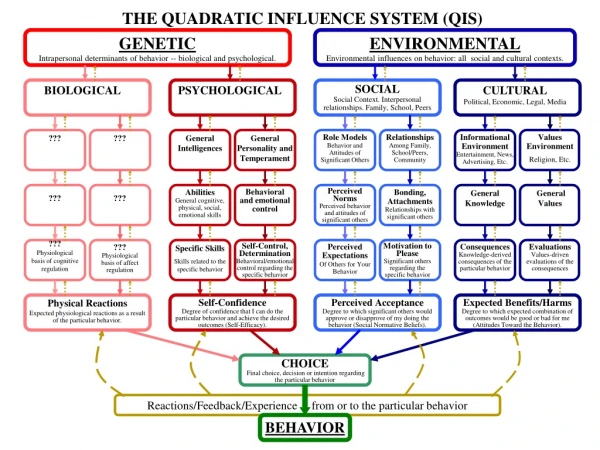
Genetic Determinants of Severe Brain Malformation. Jayne Duncan MRCPATH Course October 2010. Keywords. Massively parallel DNA sequencing Human Exome NimbleGen sequence capture system™ Malformations of cortical development Shared homozygosity Autozygosity mapping Consanguinity.

E N D
Genetic Determinants of Severe Brain Malformation Jayne Duncan MRCPATH Course October 2010
Keywords • Massively parallel DNA sequencing • Human Exome • NimbleGen sequence capture system™ • Malformations of cortical development • Shared homozygosity • Autozygosity mapping • Consanguinity
Brain Development • Development of of the human cerebral cortex is an orchestrated process involving: -Generation of neural progenitors in the periventricular germinal zones -Cell proliferation characterised by symmetric and asymmetric mitoses. -Followed by migration of post-mitotic neurons to their final destinations in 6 high ordered, functional specialized layers
Brain Development (2) • Understanding the molecular mechanisms guiding the processes involved in brain development is in its infancy. • Driven by the discovery of rare mutations that cause malformations of cortical development. • Malformations of cortical development – diverse group of severe structural brain disorders. • Characterized by deranged neuronal proliferation, migration or organization.
Mapping of Disease Loci • Mapping of putative monogenic forms of malformations of cortical development is limited by factors such as: -small kindred sizes and reduced reproductive fitness -locus heterogeneity -incorrect diagnosis • These factors restrict analysis to candidate gene approaches. • Massively parallel DNA sequencing technologies enable simultaneous sequencing of a huge number of DNA fragments. • Allow sequencing of the entire human genome to be performed but at a very high cost
Exome Sequencing • Exome sequencing can be used an alternative to whole genome sequencing in gene identification. • This involves targeted resequencing of all human protein coding sequences (the exome), a fraction of the genome, compared to whole genome sequencing. • Justification for use in gene identification includes : -Positional cloning focusing on protein coding sequences has proved successful in identifying genes responsible for monogenic disorders. -Many alleleic variants causing disease disrupt protein coding sequences. -Splice acceptor and donor sites are also targeted in analysis - A large number of nonsynonymous variants in the human genome are predicted to be deleterious
Exome Sequencing Method • The Nimblegen™ 2.1M human exome array is a commercial capture system that can capture about 180,000 human exons and about 550 miRNA sequences. • The method involves capturing the desired target sequences by selective hybridization. • Custom microarrays are designed to have DNA probes that represent the desired target sequences only. • A complex starting DNA is randomly fragmented and ends are filled in to generate a population of small (~500bp) blunt end fragments. • Common oligonucleotide linkers are ligated to the ends of the fragments. • The DNA is denatured and hybridized to a custom microarray; unbound sequences are removed by washing. • Desired target sequences are bound to the microarray; they are retrieved by elution. • The target sequences are amplified using linker specific primers. • The result is an enriched pool of target sequences ready for massively parallel DNA sequencing. -
Case Study: Identification of the WDR62 gene as the cause of severe cerebral cortical malformations • Whole exome sequencing has been used to identify recessive mutations in WD repeat domain 62 (WDR62) as the cause of a wide spectrum of severe cerebral cortical malformations including: • Microcephaly • Pachygyria with cotrical thickening • Hypoplasia of the corpus collosum Additional abnormalities that can present include: • Lissencephaly • Schizencephaly • Polymicrogyria • Cerebellar hypoplasia • All traits traditionally thought to be distinct entities before the identification of WDR62
Identification of WDR62 • Performed whole-exome sequencing of the index case (NG26-1) in a small consanguineous family from eastern Turkey. • A mean coverage of 44X was achieved and 94% of all bases were read more than four times, enough to identify novel variants with high specificity • Initially whole genome genotyping was performed in two affected family members that identified shared homozygous segments (each >2.5Mb), this helps to focus the search to a smaller number of candidate genes. -Patient (NG 26-1) showed failure to reach developmental milestones and microcephaly MRI images of a control subject compared with NG 26-1 (b, d) confirms the clinical diagnosis of microcephaly and shows a diffusely thickened cortex, an indistinct grey–white junction, pachgyria and underoperculization.
Analysis of Captured Exome DNA • The captured, purified and clonally amplified target Exome DNA was sequenced on the Illuminer Genome Analyzer IIx. • The sequence reads were aligned to the human genome. • Software such as Perl scripts and SAM (sequence alignment mapping)tools were used to detect mismatches, error positions, single nucleotide variation and indels. • Variants detected were annotated for novelty against dbSNP, nine personal genome databases and previous exome experiments.
Case Study Results • Two novel homozygous missense variants and one novel homozygous frameshift mutation identified within the shared homozygosity intervals • Frameshift mutation resides in WDR62 gene at 19q13.2 and is the result of a 4bp deletion in exon 31. • Sanger sequencing showed the mutation was homozygous in both affected individuals and heterozygous in both parents. • To determine if mutations in WDR62 accounted for additional malformations of cortical development whole genome genotyping performed in 30 probands from consanguineous parents with diagnosis of agyria or pachygyria. • Eight patients showed homozygosity of at least 2cM spanning the WDR62 region and one case had the same homozygous haplotype and the 4bp deletion spanning WDR62 locus as the initial case, suggesting fourth degree relatedness. • Sanger sequencing of coding region of WDR62 identified five novel homozygous mutations, 2 nonsense mutations, 1 frameshift caused by a 17bp deletion and 2 novel homozygous missense mutations in evolutionary conserved residues, predicted to be damaging by PolyPhen. • All mutations absent in 1290 Turkish and 1500 Caucasian control chromosomes (One exception, missense variant E526K heterozygous in 3 unrelated Turkish controls).
Results continued • To further evaluate the homozygous mutations sequenced WDR62 coding region in 12 consanguineous patients with non-neurological conditions who had segments of homozygosity of at least one million bps spanning the WDR62 locus, no variants identified. • Similarly only 4 novel heterozygous missense variants identified in 100 whole exomes of individuals with non-neurological conditions. • No copy number variants identified and only one deletion detected by BAC array is reported on Database of genetic variants • Whole Exome sequencing has implicated WDR62 in the pathogenesis of a spectrum of cortical abnormalities that were thought to be distinct, suggesting that these diverse features can have a common cause. • WDR62 lies in a 10 million bp interval previously identified as a microcephaly locus, MCPH2. • Whole-Exome sequencing and targeted resequencing suggests that WDR62 is the MCPH2 gene and extends the phenotype beyond microcephaly.
Autozygosity and Gene Mapping • Autozygosity is a term used to describe homozygosity for markers identical by descent. • The two alleles are both copies of one specific allele that was present in a recent ancestor. • In a consanguineous marriage both partners share some of their ancestors and so may have each inherited a copy of the same ancestral allele at a locus. • Their child may be autozygous for that allele or a second independent example of the same allele may have enetered the family at some stage in the pedigree. • Such alleles are identical by state. • The rarer the alleles are in a population, the more likely the homozygosity represents autozygosity.
Autozygosity Mapping using the Affymetrix Genome-wide SNP Array • DNA is cut with a restriction enzyme. • Universal adaptors (blue) are ligated to the fragments, allowing them to be amplified with a single pair of PCR primers. • PCR products are fragmented, labeled and hybridised to 25-mer oligos on the microarray. • Each oligo specifically hybridises to fragments containing one allele of one specific SNP.
Affymetrix Allele Detection • The Affymetrix system uses 40 probes per nucleotide position, each about 25 nucleotides long. • Probes are organized in sets of four, each having a different nucleotide at the central position. • Five quartets query the forward strand and five query the reverse strand.. • The five quartets are offset along the genomic sequence so that the variable nucleotide in the probe might be at position -2, -1, 0, +1 and +2 relative to the nucleotide being assayed. • Base calling is based on algorithms that compare the hybridization intensities of all 40 probes.
Case Study: Identification of TUBA8 Gene as the cause of Polymicrogyria with Optic Nerve Hypoplasia • Polymicrogyria (PMG) is a malformation of the cerebral cortex. • Usual gyral pattern is replaced by numerous small infoldings. • The normally six layered cortex is replaced by a four layered or unlayered cortex. • It is a hallmark of Bilateral Frontoparietal Polymicrogyria (BFPP) that results from faulty N-glycosylation of GPR56 protein, which has a role in the maintenance of pial basement membrane integrity during development of the cortex.
Identification of the TUBA8 Gene • Performed Autozygosity mapping in Four children from two consanguineous families of Pakistani origin. • Autosomal recessive inheritance considered due to consanguinity in both families. • Children presented with severe developmental delay, hypotonia, seizures, profound neurological impairment and optic nerve hypoplasia. • Neurological imaging showed a pattern of extensive PMG with a dysplastic or absent corpus callosum anabnormality of the brain stem and hypoplastic optic nerves.
Identification of the TUBA8 Gene (2) • Regions of concordant homozygosity identified in affected individuals from both families by performing a SNP-microarray-genome-wide homozygosity scan • Used the Affymetrix Genome Wide Human SNP Array 6.0. • One 7.42Mb region of concordant homozygosity identified on chromosome 22q11.2 in affected individuals. • There were 929 SNPs and 230 annotated genes within the shared minimal autozygous region. • The presence of the α8-tubulin gene (TUBA8) which is expressed in the brain was thought significant and considered a candidate gene. - dominant mutations in the α1-tubulin gene (TUBA1A) are known to cause lissencephaly associated with mild microcephaly and dysgenesis of the corpus callosum and brain stem hypoplasia. -More recently dominant mutations in TUBB2 resulting in asymmetric frontal predominant PMG with corpus collosum deysgenesis have been described
Results • Sanger sequencing in the affected individuals of the two families showed no coding region variants. • A homozygous 14bp deletion in intron 1 of TUBA8 was identified. • This segregated with the disease in both families and all obligate carriers were heterozygous for the mutation. • Mutation not found in 342 ethnically matched control chromosomes
Results continued • The 14bp deletion lies 11bp upstream of the exon 2 splice junction, eliminating a large portion of the acceptor site polypyrimidine tract. • Reverse transcriptase PCR analysis on lymphoblastoid RNA from one of the affected individuals showed the full length TUBA8 transcript was present at a greatly reduced level. • The major product present was a shortened transcript, confirmed by sequencing to lack exon 2.
Protein Expression Studies • The α8-tubulin protein participates in microtubule assembly in mammalian cells. • In situ hybridization studies in the developing mouse brain show Tuba8 is widely expressed in developing neural structures, particularly in post mitotic cortical neurons • At embryonic day 15.5-16.5 widely expressed in post mitotic cortical neurons. • However by postnatal day 8 Tuba8 is stongly expressed in the cortical layers II-III and V. • Suggests Tuba8 plays a role in cortical organisation
Fowler syndrome • In 1972 Fowler et al. described a family where five sisters were affected by a prenatally lethal disorder diagnosed between 26 and 33 weeks gestation. • The fetuses showed “bubble-like” cerebral hemispheres with no apparent gyral pattern, as well as a distinctive glomeruloid vascular proliferation (due to calcification and necrosis in white matter, brain stem, cerebellum and spinal cordin the central nervous system and retina. • Polyhydramnious and fetal akinesia deformation sequence (FADS) and muscular neurogenic atrophy were also apparent. • Fowler syndrome, proliferative vascularopathy and hydranencephaly-hydrocephaly (PVHH) or encephaloclastic proliferative vasculopathy is inherited as an autosomal recessive condition. • Less than 40 cases have been reported, possibly due to under-diagnosis as neuropathology is not available for many cases of FADS.
Gene Identification • To map and identify the gene for Fowler syndrome affected fetuses from three consanguineous families of Pakistani origin were investigated using autozygosity mapping. • A genome wide linkage scan was performed using Affymetrix 250k SNP microarrays to identify regions of homozygosity >2Mb common to all cases. • Two extended overlapping regions of homozygosity on chromosome 14 were identified (one region of 12.56Mb and another of 24.65Mb). • Further genotyping with microsatellite markers was then performed in all available family members. • Linkage to the 12.56 region was excluded by the finding of heterozygous alleles in one affected individual. • However microsatellite analysis within the 24.65Mb region revealed that the affected fetuses were homozygous and one unaffected sib was heterozygous.
Pedigrees and Linkage Analysis Results for 3 Families with Fowler syndrome
Gene Identification (2) • Comparison of the microsatellite data showed a common haplotype shared by the 3 families analysed. • The candidate region was narrowed by the SNP array data. • Mutation analysis of 7 genes within the shared region revealed a homozygous missense mutation c.1289C>G; p.Thr430Arg in exon 7 of the feline leukemia virus subgroup C cellular receptor family member 2 (FLVCR2 or C14orf58 or FLJ20371). • This was present in all affected cases in families 1-3 and was not detected in 646 control chromosomes of Asian and European descent. • Sequence alignment showed the threonine residue to be conserved down to C.elegans and the missense mutation was reported to be damaging by Polyphen and SIFT. • Mutation analysis in a further two non consanguineous families of Northern European origin showed the affected cases were compound heterozygotes for a 6bp deletion in exon 1 and a missense mutation in the first case and a nonsense mutation in exon 1 and a missense mutation in exon 3 in the second case. (missense mutations occurred in highly conserved residues. All mutations not detected in normal controls)
Protein Function • FLVCR2 is a protein with 12 predicted transmembrane domains that belongs to the major facilitator superfamily of secondary carriers. • These transport small solutes in response to changes in chemiosmotic ion gradients. However the function of FLVCR2 is unclear. • It has been suggested that the FLVCR2 transporter is specific for a calcium chelate complex and is involved in the regulation of growth and calcium metabolism, however this debatable as proliferation and motility of vascular endothelial cells which are major processes in angiogenesis and the induction of CNS vascularization are calcium-dependent events. • FLCVR2 and its paralog FLVCR1 share 60% amino acid identity across their 12 transmembrane domains. • FLVCR1 is a human exporter of heme, essential for erythropoiesis and serves as a receptor for feline leukemia virus subgroup C. • Feline leukemia virusesare pathogenic retroviruses of domestic cats that induce proliferative, degenerative and immunosuppressive disorders. • FeLV-A, the primary strain infects cells through the solute carrier family 19 member 2 (SLC19A2), while the secondary strain FeLV-C infects via the heme exporter FLVCR1 (recently an FeLV-C strain was detected that could infect via FLVCR2, indicating that FLVCR1 and 2 could be functionally similar.
References • Abdullahi et al Am J Hum Genet 85: 737-744: 2009 • Bilguvar et al Nature467: 207-211: 2010 • Mamanova et al Nature Methods 7: 111-119: 2010 • Meyer et al Am J Hum Genet 86: 471-478: 2010 • Strachan & Read. Human Molecular Genetics 4th Ed (2010)