Structural Bioinformatics Lecture: Introduction to Homology Modeling
390 likes | 443 Vues
Explore the principles of homology modeling, folding recognition, model building, loop and sidechain modeling, refinement techniques, and testing methods like the CASP experiment. Learn why homology modeling is essential in structural bioinformatics and how it works effectively. Discover the tools and approaches used, such as fold recognition, profile-based methods, and loop building strategies for different loop lengths. Gain insights into the Self-Consistent Mean-Field Sampling method and the Dead End Elimination Theorem in protein modeling.

Structural Bioinformatics Lecture: Introduction to Homology Modeling
E N D
Presentation Transcript
Structure prediction: Homology modeling Lecture 8 Structural Bioinformatics Dr. Avraham Samson 81-871
Homology Modeling • Presentation • Fold recognition • Model building • Loop building • Sidechain modeling • Refinement • Testing methods: the CASP experiment
Homology Modeling • Presentation • Fold recognition • Model building • Loop building • Sidechain modeling • Refinement • Testing methods: the CASP experiment
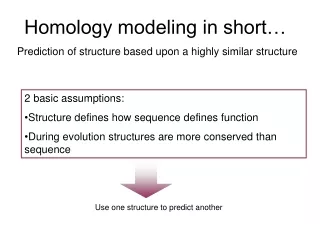
Why do we need homology modeling ? To be compared with:
Structural Genomics project • Aim to solve the structure of all proteins: this is too much work experimentally! • Solve enough structures so that the remaining structures can be inferred from those experimental structures • The number of experimental structures needed depend on our abilities to generate a model.
Structural Genomics Proteins with known structures Unknown proteins
Homology Modeling: why it works High sequence identity High structure similarity
Homology Modeling: How it works • Find template • Align target sequence • with template • Generate model: • - add loops • - add sidechains • Refine model
Homology Modeling • Presentation • Fold recognition • Model building • Loop building • Sidechain modeling • Refinement • Testing methods: the CASP experiment
Fold Recognition Homology modeling refers to the easy case when the template structure can be identified using BLAST alone. What to do when BLAST fails to identify a template? • Use more sophisticated sequence methods • Profile-based BLAST: PSIBLAST • Hidden Markov Models (HMM) • Use secondary structure prediction to guide the selection of a template, • or to validate a template • Use threading programs: sequence-structure alignments • Use all of these methods! Meta-servers: http://bioinfo.pl/Meta
Fold Recognition Blast for PDB search Full homology modeling packages Profile based approach HMM Structure-derived profiles Fold recognition and Secondary structure prediction
Homology Modeling • Presentation • Fold recognition • Model building • Loop building • Sidechain modeling • Refinement • Testing methods: the CASP experiment
Very short loops: Analytic Approach Wedemeyer, Scheraga J. Comput. Chem. 20, 819-844 (1999)
Medium loops: A database approach Scan database and search protein fragments with correct number of residues and correct end-to-end distances
Medium loops: A database approach cRMS (Ǻ) Method breaks down for loops larger than 9 Loop length
Long loops: A fragment-based approach clustered data library data 1) Clustering Protein Fragments to Extract a Small Set of Representatives (a Library)
Generating Loops Fragment library
Generating Loops Fragment library
Generating Loops Fragment library
Generating Loops Fragment library
Generating Loops Fragment library Threading
Long loops: A fragment-based approach Test cases: 20 loops for each loop length Methods:database search, and fragment building, with fragment libraries of size L <cRMS (Ǻ) Loop length
Loop building: Other methods Heuristic sampling (Monte Carlo, simulated annealing) Inverse kinematics Relaxation techniques Systematic sampling http://www.cs.ucdavis.edu/~koehl/BioEbook/loop_building.html
Homology Modeling • Presentation • Fold recognition • Model building • Loop building • Sidechain modeling • Refinement • Testing methods: the CASP experiment
i,3 i,1 i,2 Self-Consistent Mean-Field Sampling P(J,2) P(J,1) P(J,3)
i,3 i,1 i,2 Self-Consistent Mean-Field Sampling P(i,2) P(i,1)+P(i,2)+P(i,3)=1 P(i,1) P(i,3)
j,1 i,1 j,3 i,3 i,2 j,2 Self-Consistent Mean-Field Sampling Multicopy Protein
j,1 i,1 j,3 i,3 i,2 j,2 Self-Consistent Mean-Field Sampling Multicopy Protein Mean-Field Energy E(i,k) = U(i,k) +U(i,k,Backbone)
j,1 i,1 j,3 i,3 i,2 j,2 Self-Consistent Mean-Field Sampling Multicopy Protein Mean-Field Energy E(i,k) = U(i,k) +U(i,k,Backbone) Update Cycle (Koehl and Delarue, J. Mol. Biol., 239:249-275 (1994))
Self-Consistent Mean-Field Sampling Another way is simply aligning the side chains as well
Dead End Elimination (DEE) Theorem • There is a global minimum energy conformation (GMEC) for which there is a unique • rotamer for each residue • The energy of the system must be pairwise. Each residue i has a set of possible rotamers. The notation ir means residue i has the conformation described by rotamer r. The energy of any conformation C of the protein is given by: Note that:
Dead End Elimination (DEE) Theorem Consider two rotamers, irand it, at residue i and the set of all other rotamer conformations {S} at all residues excluding i. If the pairwise energy between ir and js is higher than the pairwise energy between it and js, for all js in {S}, then ircannot exist in the GMEC and is eliminated. Mathematically: If then ir does not belong to the GMEC
Dead End Elimination (DEE) Theorem This is impractical as it requires {S}. It can be simplified to: If then ir does not belong to the GMEC Iteratively eliminate high energy rotamers: proved to converge to GMEC Desmet, J, De Maeyer, M, Hazes, B, Lasters, I. Nature, 356:539-542 (1992)
Other methods for side-chain modeling • Heuristics (Monte Carlo, Simulated Annealing) • SCWRL (Dunbrack) • Pruning techniques • Mean field methods
Loop building + Sidechain Modeling: generalized SCMF Template Add multi-copies of candidate “loops” Add multi-copies of candidate side-chains Final model Koehl and Delarue. Nature Struct. Bio. 2, 163-170 (1995)
Homology Modeling • Presentation • Fold recognition • Model building • Loop building • Sidechain modeling • Refinement • Testing methods: the CASP experiment
Refinement ? CASP6 assessors, homology modeling category: “We are forced to draw the disappointing conclusion that, similarly to what observed in previous editions of the experiment, no model resulted to be closer to the target structure than the template to any significant extent.” The consensus is not to refine the model, as refinement usually pulls the model away from the native structure!!
Homology Modeling: Practical guide Approach 2: Submit target sequence to automatic servers - Fully automatic: - 3D-Jigsaw : http://www.bmm.icnet.uk/servers/3djigsaw/ - EsyPred3D: http://www.fundp.ac.be/urbm/bioinfo/esypred/ - SwissModel: http://swissmodel.expasy.org//SWISS-MODEL.html - Fold recognition: - 3D-PSSM: http://www.sbg.bio.ic.ac.uk/~3dpssm/ - Useful sites: - Meta server: http://bioinfo.pl/Meta - PredictProtein: http://cubic.bioc.columbia.edu/predictprotein/
Comparative Modeling Server & Program • COMPOSERhttp://www.tripos.com/sciTech/inSilicoDisc/bioInformatics/matchmaker.html • MODELERhttp://salilab.org/modeler • InsightIIhttp://www.msi.com/ • SYBYLhttp://www.tripos.com/