Blood Components for Health
E N D
Presentation Transcript
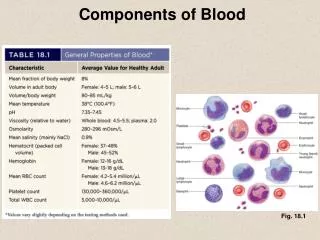
Blood Components • Plasma: • The liquid part of blood. All the blood cells are suspended in this liquid. • Contains dissolved salts (electrolytes) and proteins • Albumin helps keeps blood vessels from leaking and carries hormones and drugs to different parts of the body. • Antibodies (immunoglobulins) that defend the body against viruses, fungi, and cancer cells • Serves as a reservoir that can absorb replenish or absorb water from tissues when necessary. • Prevents blood vessels from collapsing and clotting by keeping them filled and circulating • Plays a role in warming and cooling the body
Blood Components Red Blood Cells • Erthrocytes: Make up 40% of the blood’s volume • Produced in the bone marrow • Contain hemoglobin, a protein that gives blood its red color and enables it to carry oxygen. • White Blood Cells • Leukocytes: Fewer in number than RBCs (1:660) • Primary responsibility: Defend the body against infection • Platelets • Thrombocytes: cell-like particles smaller than RBCs and WBCs. • Help with clotting process by gathering at bleeding site and clumping together to form a plug that helps seal the blood vessel.
Blood cells Granulocytes
Problems of Erythrocyte Production • Anemia – reduction of RBC volume or Hgb concentration below normal Classifications: 1. Etiology/Pathophysiology – causes of RBC/Hgb depletion 2. Morphology – changes in RBC size, shape, and color
Causes of Anemia • Nutritional deficiency – iron, folate, B12 • Increased destruction of RBCs – sickle cell anemia • Impaired or decreased rate of production – aplastic anemia • Excessive blood loss - hemophilia
Iron Deficiency Anemia • Causes - inadequate supply of iron - impaired absorption - blood loss - excessive demands for iron req’d for growth - inability for form Hgb
Iron Deficiency Anemia • Signs and Symptoms: due to tissue hypoxia > lack of energy, easy fatigability, pallor • Diagnosis: CBC with diff, red cell indices (MCV, MCH, MCHC), iron studies, physical exam • Medical Treatment: supplement with ferrous sulfate (dosages vary with age), dietary counseling
Nursing Assessment and Interventions - educate parents about nutrition - explain laboratory testing - teach parents proper administration of iron preparations, caution about high toxicity of iron
1. Sickle cell disease In sickle cell anemia (Hgb S cell disease), an abnormal gene results in production of an irregular red blood cell called hemoglobin (Hgb) S that replaces some of the normal hemoglobin A. The red blood cells collapse into a crescent shape (sickling) when stressed such as during dehydration, hypoxemia, or acidosis.
Sickle Cell Disease Pathophysiology: - vaso-occlusion from sickled RBCs , increased RBC destruction leads to the main manifestations of the disease, which include: splenic congestion and enlargement, hepatomegaly, liver failure, renal ischemia, hematuria • premature death of the cells (hemolytic anemia) • increased susceptibility to infection
Sickle Cell Disease • The frequency of one abnormal gene is the African-American population is 1 in 12 and the incidence of sickle cell anemia is 1 in 650 • The frequency of the gene is also high in Mediterranean and African populations
Sickle Cell Anemia Signs/Symptoms: • Exercise intolerance • Anorexia • Jaundiced sclera • Gallstones • Chronic leg ulcers • Growth retardation
Sickle Cell AnemiaDiagnosis • - Sickledex - Hgb electrophoresis - Stained blood smear • Vaso-occlusive crisis(resulting in necrosis) • mild to severe bone pain Humeral head infarction and osteonecrosis in a 50 year old female with sickle cell disease
pain episodes strokes increased infections leg ulcers bone damage (osteo-necrosis) yellow eyes or jaundice early gallstones lung blockage kidney damage and loss of body water in urine painful erections in men (priapism) blood blockage in the spleen or liver eye damage anemia delayed growth Complications of Sickle Cell Disease
Treatment of Sickle Cell Disease • There is no specific treatment for sickle cell disease, therefore, most therapy is supportive in treatment of the complications • Early recognition of infection, administration of prophylactic antibiotics, and vaccination may forestall or prevent other complications • If a painful crisis persists or there is infection of a major organ (brain, lung, or heart), exchange transfusion is performed to remove some of the sickle red cells - the effect is temporary
Treatment of Sickle Cell Disease • General guidelines • Taking the vitamin folic acid (folate) daily to help make new red cells • Daily penicillin until age six to prevent serious infection • Drinking plenty of water daily (8-10 glasses for adults) • Avoiding too hot or too cold temperatures • Avoiding over exertion and stress • Getting plenty of rest • Getting regular check-ups from knowledgeable health care providers
Prognosis of Sickle Cell Disease • Prognosis has improved with good supportive care, and many people with sickle cell disease survive into middle age • However, frequent admissions for painful crises, the complication of sickle cell disease, narcotic use and abuse due to chronic pain, and absence from school and work lead to significant psychological and vocational problems
Sickle Cell Anemia Nursing care: • Minimize tissue deoxygenation • Promote hydration • Minimize crises • Pain management • Administering blood transfusions • Encourage screening and genetic counseling • Parent education
Family Teaching • Teach proactive care to prevent episodes/crisis: • - Adequate fluid intake to prevent dehydration. • - Avoiding infection or early treatment. • - Moderate activity and adequate rest to avoid fatigue and hypoxia. • Early signs of impending crisis: splenic palpation to detect sequestration. • Stress need for immediate care if there are signs of crisis. • Genetic testing and counseling: • Explain that SCA is an autosomal recessive condition requiring the gene from both parents. Encourage testing of siblings to allow for childbearing planning. • Support child and family with emotional responses, grieving, and coping.
Hemophilia • Hemophilia is a group of X-linked recessive disorders that result in deficiency in one of the coagulation factors 8 or factor 9 deficiency • in the blood. X-linked recessive disorders are transmitted by carrier mothers to their sons, so usually only males are affected by hemophilia.
There are several types of hemophilia, including: • 1. Factor VIII deficiency (hemophilia A). • 2. Factor IX deficiency or Christmas disease (hemophilia B). • 3. Factor XI deficiency (hemophilia C). The most common type is hemophilia A, occurs when there is a deficiency of factor VIII in an individual. Factor VIII is essential in the activation of factor X, which is required for the • conversion of prothrombin into thrombin resulting in an inability of the platelets to be used in clot formation. • Hemophilia is classified according to the severity of the disease, ranging from mild to severe. The more severe the disease, the more likely there will be bleeding episodes
Clinical Manifestation: • 1. Prolong bleeding any where from the body, with severe factor deficiencies, circumcision, loss of deciduous teeth or a result of a slight fall or bruise. • 2. Subcutaneous or intramuscular hemorrhage is common. • 3. Hemarthrosis, bleeding into the joint cavities especially knee, ankle, elbow. • 4. Epistaxis (bleeding from the nose) • 5. Bleeding into the tissue serious if occur into the neck, mouth, or thorax. • 6. Hemorrhage anywhere the gastrointestinal tract, can lead to obstruction.
Diagnosis: • 1. History of bleeding • 2. Chromosomal studies. • 3. Blood tests, bleeding time, clothing time, PTT etc.
Nursing care: • Prevent bleeding • Recognize and control bleeding - Rest - Ice - Compression - Elevation • Prevent crippling effects of bleeding • Client education
Thalassemia: (Cooley’s anemia) • Thalassemia: (Cooley’s anemia) • Thalassemia is an autosomal-recessive disorder in which the alpha or beta polypeptide chains in hemoglobin A are impacted. Refers to those people living near the Mediterranean sea.
Thalassemia: (Cooley’s anemia) Types of thalassemia, alpha and beta. In alpha-thalassemia, synthesis of the alpha chain of the hemoglobin protein is affected. Problems with the beta chain occur more often, and the condition beta-thalassemia can be divided into three subcategories based on severity:
Thalassemia minor (also called beta-thalassemia trait): leads to mild microcytic anemia often no treatment is required. • 2. Thalassemiaintermedia: child requires blood transfusions to maintain adequate quality of life. • 3. Thalassemia major: to survive the child requires ongoing medical attention, blood transfusions, and iron removal (chelation therapy).
Pathophysiology: • In beta-thalassemia major, the beta-globulin chain in hemoglobin synthesis is reduced or entirely absent. • A large number of unstable globulin chains accumulate, causing the RBCs to be rigid and hemolyzed easily. • . An overproduction of RBCs (immature cells) may result in compensation for the hemolysis. • The increased activity causes massive bone marrow expansion and thinning of the bony cortex. • Growth retardation, pathologic fractures, and skeletal deformities (frontal and maxillary bossing) result.
Red blood cells have short life span which is easily destructed. This lead increase iron level and iron storage in the tissue called hemosiderosis. • Hemochromatosis: excessive iron storage in liver, skin, pancreas, heart, joints and testes resultant cellular damage which cause fibrosis of vital organs: heart failure, liver cirrhosis, diabetes and splenomegaly. • The excess iron is deposited in the body’s tissues, causing bronze pigmentation of the skin • If left untreated, beta-thalassemia major is fatal, but the use of chelation therapy has increased the life expectancy of these children to beyond their teen years
Signs and symptoms: • Thalassemia minor occurs as asymptomatic. • Thalassemia intermedia manifests with splenomegaly and moderate to severe anemia. • Thalassemia major (Cooley anemia) causing progressive chronic anemia: Hypoxia, headache, irritability, pallor, fatigue, and poor feeding. Bronzed complexion: iron-containing pigment may be noted due to breakdown of RBCs • If untreated, bone changes such as enlarged head and other facial changes may be noted.
Medical treatment: • keep Hgb > 9.5 g/dL. • Deferoxamine (Desferal), an iron chelating agent, with oral vitamin C may be administered to promote iron excretion • Splenectomy may be done to decrease destruction of blood cells. • Bone marrow transplantation may be done in some children. • After splenectomy, patient is at risk for infection and should receive vaccines to prevent influenza, meningitis, and pneumonia in addition to regular immunizations.
Nursing Interventions: • Support child during illness and distressing treatments. . • Monitor closely for complications of the condition and treatment: • Genetic counseling: • Encourage testing of siblings to allow for childbearing planning. • Explain that each pregnancy when both parents are carriers presents a 25% chance a child will be born with the disease and a 50% chance the child will have the thalassemia trait.
Idiopathic Thrombocytopenic Purpura(ITP) • Causes: acquired hemorrhagic disorder of unknown origin, probably an autoimmune response to disease-related antigens could be viral infection lead to produces antiplatelet antibodies. These antibodies destroy platelets. Excessive destruction of platelets results in deficiency (thrombocytopenia) leading to bleeding disorders. Which then leads to the development of petechiae, purpura, and excessive bruising. Purpura is a hemorrhagic rash in which blood collects under the tissues and they are purplish.
Idiopathic Thrombocytopenic Purpura(ITP) • Diagnosis: platelet count < 20,000, abnl bleeding time and clot retraction • Signs and Symptoms: petechiae, bruising, bleeding from mucous membranes, prolonged bleeding from abrasions • Complications include severe hemorrhage and bleeding into vital organs and intracranial hemorrhage
Medical management: • Supportive • Steroids [corticosteroides (prednisone)]. • , Anti-D antibody • Restriction of child activities. • intravenous gamma globulin increase production of platelets. • Platelet transfusions are not indicated unless a life-threatening condition such as intracranial hemorrhage is present. • Splenectomy (after 5 years of age)
Nursing Considerations • Client/Parent teaching • No contact sports • No aspirin • Prevent infection • Limits activity
Blood Transfusion Complications: • Hemolytic reactions - chills, shaking, fever - dyspnea - flank pain - progressive signs of shock • Febrile reactions • Allergic reactions - urticaria, flushing - wheezing • Circulatory overload