V11: Genetic networks
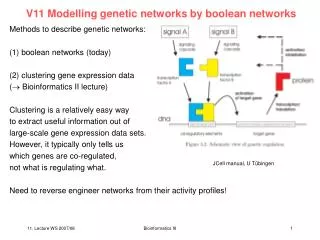
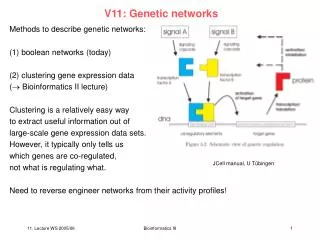
V11: Genetic networks. Methods to describe genetic networks: (1) boolean networks (today) (2) clustering gene expression data ( Bioinformatics II lecture) Clustering is a relatively easy way to extract useful information out of large-scale gene expression data sets.

V11: Genetic networks
E N D
Presentation Transcript
V11: Genetic networks Methods to describe genetic networks: (1) boolean networks (today) (2) clustering gene expression data ( Bioinformatics II lecture) Clustering is a relatively easy way to extract useful information out of large-scale gene expression data sets. However, it typically only tells us which genes are co-regulated, not what is regulating what. Need to reverse engineer networks from their activity profiles! JCell manual, U Tübingen Bioinformatics III
Intergenic interaction matrix M Since the introduction detecting gene expression by microarrays, a major problem has been the estimation of the intergenic interaction matrix M. The matrix element mijof the interaction matrix M is - positive if gene Gjactivates gene Gi - negative if gene Gjinhibits gene Gi - equal to 0 if gene Gjand gene Gi have no interaction. Gi= +1 if it is expressed, otherwise = 0. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
simulating the dynamics of regulatory networks Given the interaction matrix M, the change of state xi of gene Gibetween t and t +1 obeys a threshold rule: where H is the Heavyside function H(y) = 1 if y 0 and H(y) = 0 if y < 0, and the bi‘s are threshold values. In the case of small regulatory genetic systems, the knowledge of such a matrix M makes it possible to know all possible stationary behaviors of the organisms having the corresponding genome. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Example In the genetic regulatory network which rules Arabidopsis thaliana flower morphogenesis (right), the interaction matrix is a (11,11) matrix with only 22 non zero coefficient. Below: A fixed configuration (attractor) of its Boolean dynamics that is obtained from propagating xi(t). Mendoza, Alvarez-Buylla, JCB, 1998 Bioinformatics III
Interaction matrix - interaction graph For each genetic regulatory network, we can define an interaction graph built from the interaction matrix M by drawing an edge + (resp. -) between the vertices representing the genes j and i, iff mij > 0 (resp. < 0). To calculate the mij´s, we can either determine the s-directional correlation ij(s) between the state vector {xj(t – s)}t C of gene j at time t – s and the state vector {xi(t)}t C of gene i at time t , t varying during the cell cycle C of length K = | C | and corresponding to the observation time of the bio-array images: Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
interaction matrix and then take where is a de-correlation threshold. Alternatively, one may identify the system with a Boolean neural network. When it is impossible to obtain all the coefficients of M in this manner (either from the literature or from such calculations), it may be possible to complete M by appyling an heuristic approach. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
estimation of interaction values We may randomly choose the missing coefficients by considering - the connectivity coefficient K(M) = I / N, the ratio between the number I of interactions and the number N of genes, and - the mean inhibition weight I(M) = R / I , the ratio between the number of inhibitions R and I. For many known operons and regulation networks, K(M) is between 1.5 and 3, and I(M) between 1/3 and 2/3. If M is structurally stable, then the random estimation of M can be used to obtain an approximate estimation on the control mechanisms of the regulatory network. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Mathematical Aspects of the Inverse Problem A network with two or more connected components, i.e. two or more sub-networks, has as fixed configurations the combination (Cartesian product) of all fixed configurations of each sub-network. We say that the fixed configurations are factorizable. Thus, the inverse problem consists of determining whether a fixed configurations set is factorizable. In this way, we can obtain some information on the connectivity of the network. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Factorization Given S {0,1}n and a permutation function : {1,...,n} {1,...,n}, we denote by (S), or simply S the set {s(1)s(2) ... s(n) : s1s2...sn S }. A set S {0,1}n is said to be factorizable if there exist sets of vectors S1 {0,1}j(1) and S2 {0,1}j(2) and , ..., Sk {0,1}j(3) and a permutation function : {1, ..., n} {1,...,n} such that S can be written as S = (S1 S2 ... Sk) , where the symbol „“ is the cartesian product between sets. If S is a factorizable set, then j(1) + j(2) + ... + j(k) = n. The set defined by F = {S1,S2, ...,Sk} is called a factorization of S and each Sj F a factor of S. F is called a maximal factorization if every factor Sj F is not factorizable. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Examples i) S = {0100, 0111, 1000, 1011} = {01, 10} {00, 11}. Here, the permutation function is the identity. ii) S = {0010, 0111, 1000, 1101} = ({0100, 0111, 1000, 1011})(2,3) = ({01, 10} {00, 11})(2,3) , where (2,3) is the function which permutes the second and third coordinates. Given the sets I {1, ..., n} and S (0,1)n, let PI(S) be the projection set defined by PI(S) = {(sj(1),sj(2), ...,sj(I)): s S, j(k) I, k = 1, ..., | I |, and j(k) < j(l) for all k < l }. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Proposition 2 Proposition 2 If a set S {0,1}n is factorizable, then the maximal factorization of S is unique. Proof Let F = {S1,S2, ...,Sk} and G = {T1,T2, ...,Tk} be two distinct maximal factorizations of S. S = (S1 S2 ... Sk)1 = (T1 T2 ... Tk)2 Hence, the permutation = (1)-1○ 2 is such that S1 S2 ... Sk = (T1 T2 ... Tk) Since F and G are maximal factorizations, there is a factor of F not included into G, which is supposed to be S1 {0,1}q, q {1, ..., n}. Let T = T1 T2 ... Tm , so S1 = P{1,...,q} (T) Hence, if we denote by I(k) {1, ..., n} the set of indices such that PI(k)(T) = T, for every k = 1, ...,m and by J = { j {1,...,m}: I(j) {(1),...,(p)} } then there exists a permutation function ‘ such that Therefore, S1 is factorizable, a contradiction. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Algorithm Let : {0,1}n {0,1}n P({1,...,n}) be the function called the difference function where P({1,...,n}) is the set of subsets of {1,...,n} and defined by (x,y) = {i: xi yi}, where x,y {0,1}n. Given S {0,1}n, the idea of the Factorization algorithm is first to construct a matrix with all the values of (x,y) for every x,y S. Next, for each row i of the matrix we construct a finite and undirected graph Gi = (Vi,Ei), where the set of nodes Vi is equal to the set {1,...,n} and the set of arcs Ei is determined by the values of each row of the matrix, according to the algorithm. Finally, the connected components of the union of all graphs Gi determine the factors of the maximal factorization of S. In the case that S is not factorizable, the output of the algorithm will be a graph with a unique connected component. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Algorithm Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Theorem 3 Given a set S {0,1}n, if I = { I(1), I(2), ..., I(k) } is the output of the Factorization algorithm with input S, then F = { P(I)(S): I = 1, ..., k) is the maximal factorization of S and the complexity of the algorithm is O(|S|3 + n2) Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Example 2 Let S = { x1 = 000, x2 = 001, x3= 100, x4= 010, x5= 011, x6= 110}. The difference matrix is and the partial graphs and the output graph of the algorithm are: The output is I(1) = {1,3} and I(2) = {2}. the maximal factorization of S is given by S = (PI(1)(S) PI(2) (S))(2,3) = ({00,01,10} {0,1})(2,3) where (2,3) is the permutation of the second and third coordinates. Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Example 3 The following set of vectors corresponds to the observed fixed points of the A.thaliana regulatory network, considering only genes whose activity is not constant. Let S = { x1 = 0010000, x2 = 0011011, x3= 0000100, x4= 0001111, x5= 1100000, x6= 1101011}. The difference matrix is The graph G of the algorithm and the connected components I(1) = {1,2,3,5} and I(2) = {4,6,7} are: The maximal factorization of S is given by S = (PI(1)(S) PI(2) (S))(4,5) = ({0010,0001,1100) (000,111)}(4,5) Aracena & Demongeot, Acta Biotheoretica 52, 391 (2004) Bioinformatics III
Design principles of regulatory networks Wiring diagrams of regulatory networks resemble somehow electrical circuits. Try to break down networks into basic building blocks. Search for „network motifs“ as patterns of interconnections that recur in many different parts of a network at frequencies much higher than those found in randomized networks. Uri Alon Weizman Institute Shen-Orr et al. Nature Gen. 31, 64 (2002) Bioinformatics III
Detection of motifs Represent transcriptional network as a connectivity matrix M such that Mij= 1 if operon j encodes a TF that transcriptionally regulates operon i and Mij= 0 otherwise. Scan all n × n submatrices of M generated by choosing n nodes that lie in a connected graph, for n = 3 and n = 4. Submatrices were enumerated efficiently by recursively searching for nonzero elements. Compute a P value for submatrices representing each type of connected subgraph by comparing # of times they appear in real network vs. in random network. For n = 3, the only significant motif is the feedforward loop. For n = 4, only the overlapping regulation motif is significant. SIMs and multi-input modules were identified by searching for identical rows of M. Shen-Orr et al. Nature Gen. 31, 64 (2002) Bioinformatics III
DOR detection Consider all operons regulated by ≥ 2 TFs. Define (nonmetric) distance measure between operons k and j, based on the # of TFs regulating both operons: d(k,j) = 1/ (1+n fnMk,n Mj,n)2) Where fn = 0.5 for global TFs and fn = 1 otherwise. Cluster operons with average-linkage algorithm. DORs correspond to clusters with more than 10 connections with a ratio of connections to TFs > 2. Shen-Orr et al. Nature Gen. 31, 64 (2002) Bioinformatics III
Network motifs found in E.coli transcript-regul network a, Feedforward loop: a TF X regulates a second TF Y, and both jointly regulate one or more operons Z1...Zn. b, Example of a feedforward loop (L-arabinose utilization). c, SIM motif: a single TF, X, regulates a set of operons Z1...Zn. X is usually autoregulatory. All regulations are of the same sign. No other transcription factor regulates the operons. d, Example of a SIM system (arginine biosynthesis). e, DOR motif: a set of operons Z1...Zm are each regulated by a combination of a set of input transcription factors, X1...Xn. DOR-algorithm detects dense regions of connections, with a high ratio of connections to transcription factors. f, Example of a DOR (stationary phase response). Shen-Orr et al. Nature Gen. 31, 64 (2002) Bioinformatics III
Significance of motifs Shen-Orr et al. Nature Gen. 31, 64 (2002) Bioinformatics III
Regulatory network Each TF appears only in a single subgraph except for global TFs that can appear in several subgraphs. Shen-Orr et al. Nature Gen. 31, 64 (2002) Bioinformatics III
Structural organization of transcript-regul networks Modules: observation that reg. Networks are highly interconnected, very few modules can be entirely separated from the rest of the network. Babu et al. Curr Opin Struct Biol. 14, 283 (2004) Bioinformatics III
Evolution of the gene regulatory network Larger genomes tend to have more TFs per gene. Babu et al. Curr Opin Struct Biol. 14, 283 (2004) Bioinformatics III
Cross-organism comparison Many TF families are specific to individual phylogenetic groups or greatly expanded in some genomes. In contrast to the high level of conservation of other regulatory and signalling systems across the crown group eukaryotes, some of the TF families are dramatically different in the various lineages. Babu et al. Curr Opin Struct Biol. 14, 283 (2004) Bioinformatics III
Regulatory interactions across organisms Are regulatory interactions conserved among organisms? Apparently yes. Orthologous TFs regulate orthologous target genes. As expected, the conservation of genes and interaction is related to the phylogenetic difference between organisms. Above: Many interactions of (a) can be mapped to pathogenetic Pseudomonas aeruginosa that is related to E.coli (b). Very few interactions can be mapped from (a) to (c). Babu et al. Curr Opin Struct Biol. 14, 283 (2004) Bioinformatics III
Regulatory interactions across organisms • Observation: there is no bias towards conservation of network motifs. • Regulatory interactions in motifs are lost or retained at the same rate as the other interactions in the network. • The transcriptional network appears to evolve in a step-wise manner, with loss and gain of individual interactions probably playing a greater role than loss and gain of whole motifs or modules. Observation: TFs are less conserved than target genes, which suggests that regulation of genes evolves faster than the genes themselves. Babu et al. Curr Opin Struct Biol. 14, 283 (2004) Bioinformatics III
Analysis of complexome during cell cycle Most research on biological networks has been focused on static topological properties, describing networks as collections of nodes and edges rather than as dynamic structural entities. Here this study focusses on the temporal aspects of networks, which allows us to study the dynamics of protein complex assembly during the Saccharomycescerevisiae cell cycle. The integrative approach combines protein-protein interactions with information on the timing of the transcription of specific genes during the cell cycle, obtained from DNA microarray time series shown before. a quality-controlled set of 600 periodically expressed genes, each assigned to the point in the cell cycle where its expression peaks. Ulrik Lichtenberg Peer Bork Science 307, 724 (2005) Bioinformatics III
Temporal protein interaction network in yeast cell cycle Cell cycle proteins that are part of complexes or other physical interactions are shown within the circle. For the dynamic proteins, the time of peak expression is shown by the node color; static proteins are represented as white nodes. Outside the circle, the dynamic proteins without interactions are positioned and colored according to their peak time. Science 307, 724 (2005) Bioinformatics III
Just-in-time synthesis vs. just-in-time-assembly Transcription of cell cycle–regulated genes is generally thought to be turned on when or just before their protein products are needed: often referred to as just-in-time synthesis. Contrary to the cell cycle in bacteria, however, just-in-time synthesis of entire complexes is rarely observed in the network. The only large complex to be synthesized in its entirety just in time is the nucleosome, all subunits of which are expressed in S phase to produce nucleosomes during DNA replication. Instead, the general design principle appears to be that only some subunits of each complex are transcriptionally regulated in order to control the timing of final assembly. Science 307, 724 (2005) Bioinformatics III
Something spectacular at the end Integrate transcriptional regulatory information and gene-expression data for multiple conditions in Saccharomyces cerevisae. 5 conditions cell cycle sporulation diauxic shift DNA damage stress response Sarah Teichmann Mark Gerstein Luscombe, Babu, … Teichmann, Gerstein, Nature 431, 308 (2004) Bioinformatics III
SANDY: topological measures + network motifs + some post-analysis Luscombe et al. Nature 431, 308 (2004) Bioinformatics III
Dynamic representation of transript. regul. network a, Schematics and summary of properties for the endogenous and exogenous sub-networks. b, Graphs of the static and condition-specific networks. Transcription factors and target genes are shown as nodes in the upper and lower sections of each graph respectively, and regulatory interactions are drawn as edges; they are coloured by the number of conditions in which they are active. Different conditions use distinct sections of the network. c, Standard statistics (global topological measures and local network motifs) describing network structures. These vary between endogenous and exogenous conditions; those that are high compared with other conditions are shaded. (Note, the graph for the static state displays only sections that are active in at least one condition, but the table provides statistics for the entire network including inactive regions.) Luscombe, Babu, … Teichmann, Gerstein, Nature 431, 308 (2004) Bioinformatics III
Interpretation Half of the targets are uniquely expressed in only one condition; in contrast, most TFs are used across multiple processes. The active sub-networks maintain or rewire regulatory interactions, over half of the active interactions are completely supplanted by new ones between conditions. Only 66 interactions are retained across ≥ 4 conditions. They are always „on“ and mostly regulate house-keeping functions. The calculations divide the 5 condition-specific networks into 2 categories: endogenous and exogenous. Endogenous processes are multi-stage, operate with an internal transcriptional program Exogenous processes are binary events that react to external stimuli with a rapid turnover of expressed genes. Luscombe et al. Nature 431, 308 (2004) Bioinformatics III
Figure 2 Newly derived 'follow-on' statistics for network structures. a, TF hub usage in different cellular conditions. The cluster diagram shades cells by the normalized number of genes targeted by TF hubs in each condition. One cluster represents permanent hubs and the others condition-specific transient hubs. Genes are labelled with four-letter names when they have an obvious functional role in the condition, and seven-letter open reading frame names when there is no obvious role. Of the latter, gene names are red and italicised when functions are poorly characterized. Starred hubs show extreme interchange index values, I = 1. b, Interaction interchange (I) of TF between conditions. A histogram of I for all active TFs shows a uni-modal distribution with two extremes. Pie charts show five example TFs with different proportions of interchanged interactions. We list the main functions of the distinct target genes regulated by each example transcription factor. Note how the TFs' regulatory functions change between conditions. c, Overlap in TF usage between conditions. Venn diagrams show the numbers of individual TFs (large intersection) and pair-wise TF combinations (small intersection) that overlap between the two endogenous conditions. Luscombe et al. Nature 431, 308 (2004) Bioinformatics III
Interpretation Most hubs (78%) are transient = they are influential in one condition, but less so in others. Exogenous conditions have fewer transient hubs (different ). „Transient hub“: capacity to change interactions between connections. Luscombe et al. Nature 431, 308 (2004) Bioinformatics III
TF inter-regulation during the cell cycle time-course a, The 70 TFs active in the cell cycle. The diagram shades each cell by the normalized number of genes targeted by each TF in a phase. Five clusters represent phase-specific TFs and one cluster is for ubiquitously active TFs. Both hub and non-hub TFs are included. b, Serial inter-regulation between phase-specific TFs. Network diagrams show TFs that are active in one phase regulate TFs in subsequent phases. In the late phases, TFs apparently regulate those in the next cycle. c, Parallel inter-regulation between phase-specific and ubiquitous TFs in a two-tiered hierarchy. Serial and parallel inter-regulation operate in tandem to drive the cell cycle while balancing it with basic house-keeping processes. Luscombe et al. Nature 431, 308 (2004) Bioinformatics III
Summary • Integrated analysis of transcriptional regulatory information and condition-specific • gene-expression data; post-analysis, e.g. • Identification of permanent and transient hubs • interchange index • overlap in TF usage across multiple conditions. • Large changes in underlying network architecture • in response to diverse stimuli, TFs alter their interactions to varying degrees, thereby rewiring the network • some TFs serve as permanent hubs, most act transiently • environmental responses facilitate fast signal propagation • cell cycle and sporulation proceed via multiple stages • Many of these concepts may also apply to other biological networks. Luscombe et al. Nature 431, 308 (2004) Bioinformatics III