Statistical Tools for Microarray Experiments: Synthesizing Differentially Expressed Features
Explore how to assess common differentially expressed genes in two related experiments using statistical tools in R. This study evaluates ratios, associations, and simulation results to detect genetic patterns. Application includes analyzing mechanical ventilation effects on lung gene expression.


Statistical Tools for Microarray Experiments: Synthesizing Differentially Expressed Features
E N D
Presentation Transcript
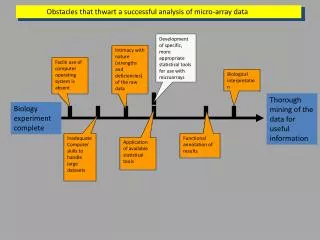
1 0 0.05 0.1 STATISTICAL TOOLS FOR SYNTHESIZING LISTS OF DIFFERENTIALLY EXPRESSED FEATURES IN MICROARRAY EXPERIMENTS Marta Blangiardo and Sylvia Richardson1 1 Centre for Biostatistics, Imperial College,St Mary’s Campus, Norfolk Place London W2 1PG, UK. m.blangiardo@imperial.ac.uk SCOPE OF THE WORK Consider two different but related experiments, how to assess whether there are more differentially expressed genes in common than expected by chance? All the computations have been performed in R and are available on BGX website (www.bgx.org.uk) R(q*)=1.0 q*=0.05 O11(q*)=18 RANKED LISTS Suppose we have two experiments, each reporting a measure (e.g. p-value,…) of differential expression on a probability scale: Small p value: MOST differentially expressed Large p value: NOT differentially expressed O1+(q) O+1(q) SIMULATION We use three batches of simulations differing by level of association between experiments and percentage of DE genes. For every batch we simulate two lists of 2000 p-values (Allison et al.2002) averaging the results over 100 simulations. We rank the genes according to the probability measures. For each cut off q we obtain a 2X2 table: The number of genes in common by chance is The number of genes observed in common is O11(q) RATIO We propose to calculate the maximum of the observed to expected ratio: It is the maximal deviation from the underneath independence model. NO association is declared when the two lists are not associated (MC p-value not significant, CI include 1) When there is a TRUE association: Conditional Model Joint Model • Increasing • % of • DE genes • The ratio T(q*) decrease • q*, O1+(q), O+1(q), and O11(q) increase • MC p-value is more significant • The ratio R(q*) decreases • q*, O1+(q), O+1(q), and O11(q) increase • CI95 are narrower R(q*) is always smaller than T(q*) and its q* is slightly bigger as it accounts for the additional variability • By using the maximum ratio, multiple testing issues for different list sizes are avoided • Returns a single list of O11(q) genes for further biological investigation • APPLICATION: analysis of deleterious effect of mechanical ventilation on lung gene expression We re-analyse the experiment presented in Ma et al, 2005, investigating the deleterious effect of mechanical ventilation on lung gene expression through a model of mechanical ventilation-induced lung injury (VILI) on rodents (mice and rats). • We analyse separately the two dataset using Cyber-T (Baldi and Long, 2001) • We use RESOURCERER to reconstruct the list of orthologs for the two species • We apply the methodology described to the lists of 2969 p-values (ortholog genes) PERMUTATION TEST Given a threshold q and fixed margins But the distribution of T(q*) is not easily obtained since the tables are nested in each other. We take advantage of the empirical distribution for T(q*) obtained via permutations. • 97 genes found in common between mice and rats • 15 genes in common with the original analysis (which highlighted 48 genes) • Two enriched pathways with our methodology: • 1) MAPK signalling activity. 6 out of the significant orthologs are involved in this KEGG pathway (Fgfr1, Gadd45a, Hspa8, Hspa1a, Il1b, Il1r2) while only 4 were highlighted in the original one. • 2) Cytokine-Cytokine receptor interaction. 5 out of the significant orthologs are involved in this KEGG pathway (IL6, Il1b, Il1r2, CCL2, Kit) while only 4 were highlighted in the original one. We perform a Monte Carlo test of T under the null hypothesis of independence between the two experiments using permutations. This returns a Monte Carlo p-value. Not associated P-value 0.8 Associated P-value <0.001 R(q*) = 1.43 q* = 0.01 CI95 = [1.13-1.75] • LIMITATIONS OF THE TEST • The uncertainty of the margins is not taken into account • The size of the list of genes in common can be vary small (typically when the total number of DE genes is small) and this can cause an instability in the estimate of T(q) • We propose a Bayesian model treating also the margins as random variables T(q*)=1.44 q*=0.01 MC p-value <0.001 O11(q*) = 97 O1+(q*) = 393 O+1(q*) = 886 BAYESIAN MODEL Starting with the 2x2 table we specify a multinomial distribution of dimension 3 for the vector of joint frequencies: • DISCUSSION • This is a simple procedure to evaluate if two (or more) experiments are associated • The permutation test gives a first look under the model where the marginal frequencies are fixed • The Bayesian model permits to enlarge the scenario introducing variability on all the components • It is very flexible and adaptable for comparisons of several experiments at different levels (gene level, biological processes level) and for different problems (e.g. comparison between species, comparison between platforms) and the vector of parameters q is modelled as non informative Dirichlet: q ~ Di(0.05,0.05,0.05,0.05) The derived quantity of interest is the ratio of the probability that a gene is in common to the probability that a gene is in common by chance: Since the model is conjugated the posterior distribution for q is Dirichlet REFERENCES Allison et al. (2002), “A mixture model approach for the analysis of microarray gene expression data”, Computational Statistics And Data Analysis, 39, 1-20. Baldi and Long, (2001) “A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes”, Bioinformatics, 17, 509-519. Ma et al., (2005) “Bioinformatics identification of novel early stress response genes in rodent models of lung injury”, Am J Physiol Lung Cell Mol Physiol 289(3), 468-477. DECISION RULE We can obtain a sample from the posterior distribution of the derived quantities R(q) and calculate the credibility interval (CI) at 95% for each threshold q. We define q* as the value of the argument for which the median of R(q) attains its maximum value, only for the subset of credibility intervals which do not include 1: Then R(q*) is the ratio associated to q*. ACKNOWLEDGEMENTS We would like to thank Natalia Bochkina, Alex Lewin and Anne-Mette Hein for helpful discussions. This work has been supported by a Wellcome Trust Functional Genomics Development Initiative (FGDI) thematic award ``Biological Atlas of Insulin Resistance (BAIR)", PC2910_DHCT