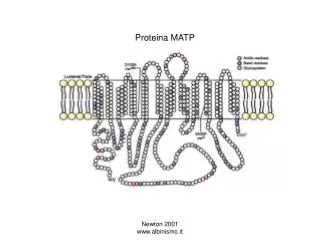
Proteina C
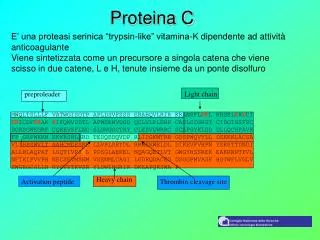
Proteina C. E’ una proteasi serinica “trypsin-like” vitamina-K dipendente ad attività anticoagulante Viene sintetizzata come un precursore a singola catena che viene scisso in due catene, L e H, tenute insieme da un ponte disolfuro. preproleader. Light chain.

Proteina C
E N D
Presentation Transcript
Proteina C E’ una proteasi serinica “trypsin-like” vitamina-K dipendente ad attività anticoagulante Viene sintetizzata come un precursore a singola catena che viene scisso in due catene, L e H, tenute insieme da un ponte disolfuro preproleader Light chain MWQLTSLLLF VATWGISGTP APLDSVFSSS ERAHQVLRIR KRANSFLEEL RHSSLERECI EEICDFEEAK EIFQNVDDTL AFWSKHVDGD QCLVLPLEHP CASLCCGHGT CIDGIGSFSC DCRSGWEGRF CQREVSFLNC SLDNGGCTHY CLEEVGWRRC SCAPGYKLGD DLLQCHPAVK FPCGRPWKRM EKKRSHLKRD TEDQEDQVDP RLIDGKMTRR GDSPWQVVLL DSKKKLACGA VLIHPSWVLT AAHCMDESKK LLVRLGEYDL RRWEKWELDL DIKEVFVHPN YSKSTTDNDI ALLHLAQPAT LSQTIVPICL PDSGLAEREL NQAGQETLVT GWGYHSSREK EAKRNRTFVL NFIKIPVVPH NECSEVMSNM VSENMLCAGI LGDRQDACEG DSGGPMVASF HGTWFLVGLV SWGEGCGLLH NYGVYTKVSR YLDWIHGHIR DKEAPQKSWA P Heavy chain Activation peptide Thrombin cleavage site
Attivazione e funzione della Proteina C L’enzima viene attivato dalla trombina che taglia il peptide di attivazione all’estremità N-terminale della catena H. Questa reazione avviene sulla parete delle cellule endoteliali ed è catalizzata dalla trombomodulina. Il recettore per la proteina C (EPCR) stimola la sua attivazione legando e presentando la proteina al complesso trombina-trombomodulina La proteina C attivata (APC) inattiva mediante taglio proteolitico i fattori procoagulanti Va e VIIIa. Queste reazioni sono catalizzate dai cofattori della APC: Proteina S e fattore Vac. Il fattore V intatto tagliato proteoliticamente dalla APC negli stessi siti in cui essa inattiva il fattore Va, diventa il fattore Vac, con attività anticoagulante.
Attivazione e funzione della Proteina C ac From: B.Dahlback and B.O.Villoutreix Journal of Thrombosis and Haemostasis, 1: 1525–1534
The 2.8 A crystal structure of Gla-domainless activated protein C (T.Mather and C.Esmon, The EMBO Journal vol.15 no.24 pp.6822-6831, 1996) EGF-like calcium binding domain EGF-like domain PPACK Active site H57,D102,S195 Catalytic domain
Mutazioni nella sequenza possono portare a carenza di proteina C, un disturbo associato al rischio di trombofilia. Si riconoscono due tipi di carenza tipo I concentrazione e funzione ridotte tipo II normale concentrazione ridotta funzione tipo II b ridotta solo attività anticoagulante tipo II a ridotte le attività anticoagulante e amidolitica
Mutazioni Sostituzione amino acidica PDB Swiss-Prot Tipo di carenza di PC Positione nel gene Precedentemente descritta 1 R-11C NP 32 I Esone 1 Si 2 E29stop H66N I171T NP 66 17 71 108 213 I Esone 2+4+6 No 3 L55Stop 55 97 I Esone 4 No 4 F76L 76 118 I Esone 4 Si 5 C78Stop 78 120 I Esone 4 Si 6 C98R 98 140 I Esone 5 No 7 R169Q NP 211 I Esone 6 Si 8 Q184L 30 226 I Esone 6 No Mutazioni in pazienti con carenza di Proteina C
Localizzazione dei mutanti sulla sequenza della proteina C
Mutazioni che mappano sulla struttura cristallizzata della proteina
Scopo I • Interpretare a livello molecolare gli effetti indotti da mutazioni missenso sulla struttura della proteina • Formulazione di ipotesi riguardo la relazione tra mutazione e carenza di proteina
Modelling dei mutanti in Insight II • Sostituzione del residuo nativo con quello mutato • Scelta del rotamero a minor energia • Minimizzazione energetica della struttura mutata
Mutazioni nella catena leggera • C98R: rottura ponte S-S tra C98 e C109 • Dominio EGF-like • Carenza di tipo I: • Alterazione del processo di folding della proteina Proteina nativa Mutante C98R C98 R98 C109 C109 Ponte S-S
Mutazioni nella catena leggera • F76L • EGF-like calcium binding: la F76 è parte di una • sequenza consenso di legame allo ione Ca++, • necessario alle interazioni tra la proteina C e i • suoi cofattori • E’ estremamente conservata tra le proteasi • seriniche umane e eventualmente sostituita da • un altro residuo aromatico • Carenza di tipo I
Modello di interazione Trombina F76 Proteina C Trombomodulina From: Fuentes-Prior PNature 404:518-25 (2000.)
Conservazione dei residui mutati all’interno di un allineamento multiplo della catena leggera della proteina C con altre proteasi seriniche umane 76 98
Mutazioni nella catena pesante G43E, D194N e G216D: sono localizzate in prossimità del sito attivo, determinano la perdita o l’acquisto di un residuo carico e riguardano residui altamente conservati
Distanze dell’ossigeno carbossilico dei residui D216, E43 e D194 dall’azoto ND1 dell’istidina catalitica 57 D216 10.85A D194 9.8A H57 E43 10.77A
D194N • Appartiene al pattern delle proteasi seriniche di PROSITE • 194 195 • [DNSTAGC] - [GSTAPIMVQH] - x(2) - G - [DE] - S – • G - [GS] - [SAPHV] - [LIVMFYWH] - [LIVMFYSTANQH] • dove precede la serina catalitica • E’coinvolto in un legame ionico interno alla proteina con la • leucina 16 N-terminale • Associato a una carenza di tipo II G43E • Associato a una carenza di proteina C di tipo I
G216D D216 G216 D189 G226 Glicina 216 si trova nella tasca di specificità per il substrato delle tripsine E’ associato a una carenza di proteina C di tipo II
G216D Da un’analisi più accurata dell’allineamento multiplo della proteina C, si si è individuata una sequenza che in posizione 216 porta proprio un aspartico: si tratta della triptasi a-1. E’ una proteasi serinica “trypsin-like” contenuta nei mastociti, che risulta praticamente inattiva se comparata alla sua omologa triptasi 2, che porta una glicina in posizione 216. La sovrapposizione delle strutture cristallografiche delle due triptasi, rivela che le differenze maggiori risiedono nel segmento 214-220, responsabile del legame con il substrato.
G216D Verde = tripasi b Rosa = loop 214-220 della tripasi a D189 G226 D216 Sarebbe proprio la presenza dell’aspartico in posizione 216 a causare una distorsione del segmento 214-220, la quale altera la conformazione del sito di riconoscimento del substrato e impedisce la presentazione corretta ai residui catalitici del legame peptidico da tagliare. E nella proteina C?
Distribuzione del potenziale elettrostatico mappato sulla superficie molecolare (Delphi) Native H57 H57 D216 G216 Neutralizzazione parziale della carica positiva dell’istidina catalitica
Distribuzione del potenziale elettrostatico mappato sulla superficie molecolare (Delphi) G43 E43 H57 H57 Neutralizzazione parziale della carica positiva dell’istidina catalitica
Distribuzione del potenziale elettrostatico mappato sulla superficie molecolare (Delphi) Native H57 H57 N194 D194 Neutralizzazione generalizzata del sito attivo e aumento della regione di positività intorno all’istidina catalitica
Una variazione dello stato di protonazione dell’istidina può interferire in due punti del meccanismo catalitico Una variazione del potenziale elettrostatico nel sito attivo implica una variazione della pKa dei gruppi titolabili
Mutant Residue Atom potential (KT/e) DpKa Asp194Asn Asn Od1 1.6184 -0.695 Asp194Asn Asn Od2 1.5863 Gly43Glu Glu Oe1 2.3166 0.91 Gly43Glu Glu Oe2 1.8911 Gly216Asp Asp Od1 3.1057 1.066 Gly216Asp Asp Od2 1.8051 Abbiamo calcolato lo shift della pKa dell’istidina 57 nei 3 mutanti (Delphi) ФO eФmrappresentano il valore medio dei potenziali elettrostatici (calcolati sui due ossigeni carbossilici del residuo acido coinvolto nella mutazione) rispettivamente nella proteina nativa e nel mutante γi = -1 perché si tratta di gruppi acidi
D194N G216D G43E In tutti e 3 i mutanti ipotizziamo un’alterazione dello stato ionico Asp102(-) His57(+) Ossianione(-) della struttura tetraedrica dello stato di transizione durante la reazione catalitica. La destabilizzazione degli stati di transizione riduce l’attività enzimatica
Scopo II • Raccolta dei mutanti da noi analizzati e dei risultati • prodotti in una banca dati • Completamento della banca dati con mutanti • della proteina C già descritti e presenti in risorse • online come: • la banca dati di varianti di Swiss-Prot • il database HGMD (Human Gene Mutation Database) • una banca dati di varianti della proteina C non più • aggiornata dal 1995
Organizzazione della banca dati PROCMD: Protein C Mutations Database
Scopo III • Rendere tutti i dati raccolti nella banca dati accessibili • al pubblico gratuitamente tramite un’interfaccia web • Permettere all’utente di manipolare ed indagare i modelli • tridimensionali dei mutanti grazie a un linguaggio di • programmazione grafica • Offrire all’utente la possibilità di modellare un proprio • mutante non contenuto nel database grazie a un programma • automatico di modelling comparativo • Creare una risorsa web altamente specialistica in quanto • dedicata ad un’unica proteina
3D Protein C Mutations Database: la home page www.itb.cnr.it/procmd
Ricerca di un mutante contenuto nel database: • di un residuo o di un intervallo di residuiper posizione • per sostituzione amminoacidica • per parole chiave o testo • per domini della proteina • estrazione di tutti i record presenti nel database Esempio di ricerca: tutti i mutanti contenuti nell’intervallo di sequenza 200-220 (secondo la numerazione PDB)
I risultati della ricerca vengono visualizzati raccolti in una tabella. Ad ogni mutante è associato un codice identificativo univoco. Esempio: selezioniamo la variante180 come indicato dalla freccia
Per ogni variante sono presenti 4 gruppi di informazioni organizzati in un’unica pagina web. 1) Informazioni generali 2) Eventuali riferimenti bibliografici del mutante e i relativi collegamenti alla banca dati PubMed
4) Una galleria di immagini tridimensionali Per la proteina nativa: -un file di coordinate PDB -un file in linguaggio VRML Per la proteina mutante: -un file di coordinate PDB -un file in linguaggio VRML Risultati dell’analisi della distribuzione del potenziale elettrostatico sulla superficie molecolare
Il file VRML Zoommare Traslare Ruotare
A ciascuna immagine della galleria è associata una pagina di commenti e informazioni, accessibile selezionando “read 3d notes”: Le informazioni riguardano: -il tipo di sostituzione -gli eventuali legami a idrogeno in cui sono coinvolti il residuo normale e mutato -le eventuali interazioni idrofobiche -la possibile interazione del residuo con il complesso trombina-trombomodulina sulla base di studi di letteratura -informazioni accessorie derivanti dai nostri studi
Modellare il proprio mutante Visualizzare la conservazione del residuo di interesse nell’allineamento multiplo con altre proteasi seriniche Ottenere le coordinate della struttura del mutante modellato attraverso Modeller, un tool automatico di modelling comparativo
Ringraziamenti Dott. Luciano Milanesi ITB-CNR Dott.ssa Ermanna Rovida ITB-CNR Dott.ssa Pasqualina D’Ursi ITB-CNR Dott. Andrea Caprera ITB-CNR E tutti i ragazzi del laboratorio di bionformatica dell’ITB- CNR dell’Istituto LITA di Segrate (MI).
DpKa = pKa (mutante) – pKa (nativa) • D194N • DpKa < 0 => pKa • 2) G43E e G216D • DpKa > 0 => pKa