Download

1 / 49
690 likes | 1.92k Vues
Oxidation of fatty acids and K etogenesis. Dr.S.Chakravarty M.D. Specific Learning Objectives . At the end of this lecture students will be able to :-

E N D
Oxidation of fatty acids and Ketogenesis Dr.S.ChakravartyM.D.
Specific Learning Objectives • At the end of this lecture students will be able to :- • Describe the role of carnitine in fatty acid transport across mitochondria for oxidation and list the fatty acids which do not require carnitine for transport. • Describe the functions of Carnitine and its deficiency due to primary and secondary causes. • Differentiate the clinical features of CPT-1 and CPT-2 deficiency • Differentiate the various types of oxidation of fatty acids and its importance • Describe the clinical features of MCAD deficiency and its treatment • Describe Zellweger syndrome and Jamaican vomiting sickness
Triglycerides + Hormone sensitive lipase Glucagon, Epinephrine Glycerol Fatty acids Glycerol kinase Beta oxidation Glycerol-3-PO4 Acetyl Co-A Ketone body synthesis Liver Liver Any cell with mitochondria Gluconeogenesis Glucose Mobilization of triglycerides from adipose tissue
Biomedical Importance • Fatty acid oxidation is not the simple reverse of fatty acid biosynthesis but an entirely different process taking place in a separate compartment of the cell. • Its an aerobic process requiring oxygen. • Increased fatty acid oxidation is a characteristic of starvation and of diabetes mellitus, leading to ketone body production by the liver (ketosis).Ketone bodies are acidic and when produced in excess over long periods, as in diabetes, cause ketoacidosis, which is ultimately fatal.
Gluconeogenesis is dependent upon fatty acid oxidation, any impairment in fatty acid oxidation leads to hypoglycemia. This occurs in various states of carnitine deficiency or deficiency of essential enzymes in fatty acid oxidation, eg, carnitinepalmitoyltransferase, or inhibition of fatty acid oxidation by poisons, eg, hypoglycin.
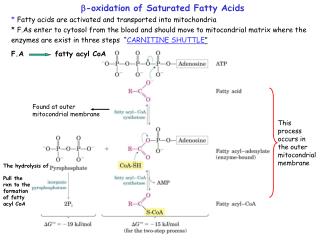
Definition: - The process in which two carbons are cleaved at a time from acyl-CoA molecules starting at the carboxyl end. The chain is broken between the α(2)- and β(3)-carbon atoms—hence the name β-oxidation. • Tissue : Liver , adipose tissue , skeletal muscles • Subcellular site :- Mitochondrion • Transport of fatty acids in Blood :- In plasma, UFA (unesterified fatty acids ) or (FFA) free fatty acids are combined with albumin. • Inside the cell they are attached to fatty acid binding protein or Z-binding protein. So infact they are never free.
Three Stages of Beta-Oxidation • Activation of fatty acids. • Transport of activated fatty acids into mitochondria. 3 Oxidation of fatty acids.
Step 1 : Activation of fatty acids FFA + ATP +CoA acyl CoA AMP + PPi + Acyl CoA synthetase PPi + H2O inorganic 2Pi pyrophosphatase THIS IS THE ONLY STEP THAT REQUIRES ATP
Step2. Long chain fatty acids penetrate inner mitochondrial membrane as carnitine derivatives Carnitine :--hydroxy trimethyl ammonium butyrate
Acyl CoA dehydrogenase chain 2 Enoyl CoA Hydratase L(+)-3-hydroxy acyl CoA dehydrogenase Thiolase + Step 3. Beta oxidation of fatty acids
Energetics of Beta oxidation Eg. Palmitic acid (16 C) Needs 7 cycles to produce 8 Acetyl CoA
Regulation of fatty acid oxidation: Fatty acyl Co-A Malonyl Co-A Cytoplasm (-) Carnintine acyl Transferase -1 (CAT or CPT) Outer mitochondrial membrane Mitochondrial matrix Rate limiting step
-oxidation • Detected in Brain • Removal of one carbon at a time from the carboxy end of the molecule. • Alpha-oxidation of phytanic acid is believed to take place entirely within peroxisomes. • No requirement of CoA derivatives • Does not generate high energy phosphates - oxidation very minor pathway and is brought about by hydroxylases of cyt.P450 in E.R. The –CH3 group is converted to –CH3OH and finally oxidized to –COOH , thus forming a dicarboxylicacid.This is -oxidized usually to adipic(C6) or suberic acids(C8) , which are excreted in the urine.
Very long chain fatty acid >20 carbon Step wise process Peroxisomes Very long chain acyl Co-A dehydrogenase Long chain fatty acid 12- 20 carbon Long chain acyl Co-A dehydrogenase Medium chain fatty acid 6-12 carbon Mitochondria Medium chain acyl Co-A dehydrogenase Short chain fatty acid <6 carbon Short chain acyl Co-A dehydrogenase Acetyl Co-A
Peroxisomes • A modified form of oxidation is found in peroxisomes and leads to the formation of acetyl-CoA and H2O2 (from the flavoprotein-linked dehydrogenase step), which is broken down by catalase. • Thus, this dehydrogenation in peroxisomes is not linked directly to phosphorylation and the generation of ATP. These enzymes are induced by high-fat diets and in some species by hypolipidemic drugs such as clofibrate. • The enzymes in peroxisomes do not attack shorter-chain fatty acids; the -oxidation sequence ends at octanoyl-CoA. • Octanoyl and acetyl groups are both further oxidized in mitochondria. • Another role of peroxisomal oxidation is to shorten the side chain of cholesterol in bile acid formation . • Peroxisomes also take part in the synthesis of ether glycerolipids, cholesterol, and dolichol.
Oxidation of odd chain fatty acids • Oxidation of fatty acid with odd no. of carbon atom yields acetyl CoA + Propionyl CoA( Enters TCA as Succinyl Co-A).Thus this portion of odd chain fatty is glucogenic.
Methylmalonicaciduria • Deficiency of vitamin B12 – accumulation of methyl malonyl Co-A. • Mutation in methyl malonyl Co-A mutase • Converted to Methyl malonic acid. • Methylmalonicacidemia and later methylmalonicaciduria. • Metabolic acidosis.
Oxidation of unsaturated fatty acids: • Lower energy yield compared to saturated fatty acids of same number of carbon atoms. • Also occurs in mitochondria with little modification of few enzyme steps.
Fat is burnt on the wick of carbohydrates • Oxidation of fats need the help of Oxaloacetate which enters into the cycle and is regenerated in the end . • The major source of OAA is Pyruvate. (Carbohydrate)
Clinical Aspects • Impaired oxidation of fatty acids gives rise to diseases associated with nonketotic hypoglycemia . • Carnitine deficiency can occur in preterm babies owing to inadequate biosynthesis or renal leakage. • t/t is by giving oral carnitine supplements • CPT 1 deficiency affects only the liver – hypoglycemia • CPT2 deficiency affects primarily skeletal muscle and only when severe the skeletal muscle .
Medium chain acyl Co-A dehydrogenase deficiency (MCAD) • Most common inborn error of fatty acid metabolism • Defect in first step of beta oxidation • Present in milk – seen in infants dependent on mother’s milk. • Accumulation of medium chain fatty acids.
Cont… • Alternate omega oxidation – leads to accumulation of dicarboxylic acids adipic (C6), suberic (C8), sebacic (C10). • Dicarboxylicaciduriaandpresence of medium chain acyl carnitines in urine • Profound Hypoglycemia – no ATPs for running gluconeogenesis. • Absent ketone bodies in the bloodwhy ?no acetyl Co-A • Hyperammonemia- octanoic acid mitochondrial poison coma. • Common cause of sudden infant death syndrome (SIDS)
Carnitine deficiency • Newborn infants esp PRETERM –Inadequate biosynthesis and renal leakage. • Hypoglycemia . • Oral Carnitine supplementation.
Jamaican vomiting sickness: • Unripe Ackee fruit – Hypoglycin • Inactivates medium and short chain acyl Co-A dehydrogenase. • Dicarboxylicaciduria – due to omega oxidation. • intractable vomiting, abdominal pain, and lethargy and is profoundly hypoglycemic
Zellweger syndrome • Peroxisomal disorder • Cerebrohepatorenal syndrome • Defect in peroxisomal biogenesis in all tissues. Clinical features: death by 1 year. • Impaired neuronal migration and brain development • Craniofacial dysmorphism • Renal cyst and Hepatomegaly.
Refsum’s disease • Refsum’s disease is a rare neurologic disorder caused by accumulation of phytanic acid formed from Phytol , a constituent of chlorophyll . • Susceptible people have a deficiency of phytanoyl-CoA hydroxylase that prevents -oxidation . • Phytanic acid contains a methyl group on C3 that blocks -oxidation also.
Acute fatty liver of pregnancy • Defects are known in long chain 3-hydroxyacyl-CoAdehydrogenase may be a cause of acute fatty liver of pregnancy.
A 4-month-old infant presents with a seizure. His mother reports that her infant has been irritable and lethargic over the past several days. The infant is found to be profoundly hypoglycemic and have low ketones . Short-chain dicarboxylic acids are found to be elevated in the serum. The most likely enzyme deficiency is which of the following? (A) Medium-chain acyl CoA dehydrogenase (MCAD) (B) Carnitine acyltransferase I (C) Hormone-sensitive lipase (D)Pyruvatecarboxylase (E) Fatty acyl CoAsynthetase
What is the primary role of carnitine in fatty acid oxidation ? (A) Activates long-chain fatty acids in the cytosol (B) Transport of acyl groups across the inner mitochondrial membrane (C) Is converted to enoylCoA (D) Is converted to β-hydroxyacylCoA (E) Is involved in breakdown of even-chain, but not odd-chain, fatty acids
An infant is born with a high forehead, abnormal eye folds, and deformed ear lobes and shows little muscle tone and movement. After multiple tests, he is diagnosed with Zellweger syndrome, a disorder caused by peroxisome malformation. What type of fatty acid would you expect to accumulate in patients with Zellweger syndrome? (A)Short-chain fatty acids (B) Acetyl CoA (C)Dicarboxylic acids (D) Long-chain fatty acids (E) Very-long-chain fatty acids
Ketogenesis • Ketogenesis Occurs When There Is a High Rate of Fatty Acid Oxidation in the Liver • Normal plasma ketone bodies concentration =0.2 mmol/L • Loss via urine is less than 1mg/24hrs.
Utilization in extrahepatic tissues: • Liver can only synthesize ketone bodies but it cannot utilize them. • Only extrahepatic tissues can utilize acetoacetate – because of the enzyme Succinyl-CoA-acetoacetatetransferase – (THIOPHORASE).
Transport of ketone bodies from the liver and pathways of utilization and oxidation in extrahepatic tissues.
Regulation of ketogenesis • Lipolysis in adipose tissue (as FFA are precursors of KB) • After uptake by liver FFA are oxidized to CO2 or esterified to triacylglycerol and phospholipids . • acetyl CoA formed in -oxidation is oxidized in TCA cycle or it enters the pathway of ketogenesis to form ketone bodies . Complete oxidation of 1 mol of palmitate actually produces 106mol ATP by TCA but only 26 when acetoacetate is the end product and 21 when 3-hydroxy butyrate is the end product. Thus , ketogenesis may be regarded as a mechanism that allows the liver to oxidize increasing quantity of fatty acids within a tightly coupled system of oxidative phosphorylation.
Diabetic ketoacidosis: • Mainly seen in type-1 diabetes mellitus • Hyperglycemia >300mg/dl • Dehydration – Glucosuria, ketonuria • High anion gap Metabolic acidosis (pH <7.2) – due to ketone bodies • Low Bicarbonate levels (<15mEq/L) – used for neutralization • Hyperkalemia - >5-5.5mg/dl
Diabetic ketoacidosis: • Decreased insulin glucagon ratio- increased fatty acid oxidation increased ketone bodies. • At physiological pH exists as ketoacids neutralized by bicarbonate metabolic acidosis. • Also increased excretion of ketones in urine –HCO3 also excreted to maintain urine pH.
Glucose not entering cells of SEKLETAL MUSCLES AND ADIPOSE TISSUE due to Insulin deficiency (GLUT 4 receptors ) • CELLS is actually STARVING !! • INCREASED MOBILIZATION OF LIPIDSIncreased FATTY ACID OXIDATION Increased Acetyl CoA. • Less OAA (less Pyruvate Less OAA)Less utilization of acetyl CoA in TCA cycle KETOGENESIS
Diabetic ketoacidosis: • Exacerbated by tissue hypoxia and infections. • Nausea and vomiting • Abdominal pain – due to a/c pancreatitis due to hypertriglyceridemia • Kussumal’s breathing: metabolic acidosis. • Fruity odour – acetone • Hyperglycemia – Glucosuria with water loss. • Hypotension and coma.
A 12-year-old boy presents with fatigue, polydipsia, polyuria, and polyphagia. A fingerstick glucose measurement shows a glucose level of 350mg/dL in his serum. He is diagnosed with type 1 diabetes mellitus, a disease characterized by a deficiency of insulin. Which one of the following is most likely occurring in this patient? A) Increased fatty acid synthesis from glucose in liver B) Decreased conversion of fatty acids to ketone bodies C) Increased stores of Triacylglycerol in adipose tissue D) Increased production of acetone E) Chronic pancreatitis