PART III. MACROEVOLUTION
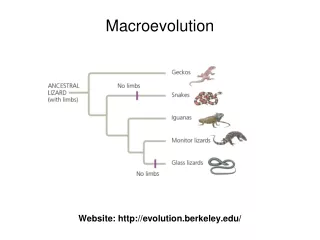
PART III. MACROEVOLUTION We already considered History of Life and learned a lot about Macroevolution that occurred in the past - but only at the shallow level of chronology and generalizations. 3b) Cladogenesis and extinction are extremely unfair processes


PART III. MACROEVOLUTION
E N D
Presentation Transcript
PART III. MACROEVOLUTION We already considered History of Life and learned a lot about Macroevolution that occurred in the past - but only at the shallow level of chronology and generalizations. 3b) Cladogenesis and extinction are extremely unfair processes Now, after studying Microevolution, we are ready to treat the same subject deeper, and to try to understand hidden mechanisms of Macroevolution. For some generalizations simple explanations may be enough, but Macroevolution is such a complex and mysterious process that it must be based on theory, which is so far absent. GENERALIZATION: New genes mostly appear from pre-existing genes - of course, this is an easy way. IN NEED OF A DEEP THEORY: Changing <1% of the genome is enough to turn an ape into a human - how? We will consider partial theories of Macroevolution at all levels, starting from sequences.
Macroevolution at different levels: At the level of sequences (genomes), Macroevolution is relatively well-understood. In contrast, Macroevolution at the next three levels - molecules, cells and organisms - is understood very poorly. However, the two upper levels - of populations and of ecosystems, are simpler again, and there are many useful partial theories of their Macroevolution. Sequences are just genetic texts - they are not doing anything directly, and are, thus, relatively easy to study. In contrast, molecules, cells and organisms are levels where real action occurs. Not surprisingly, studying them is tough. However, complexity of adaptations can be ignored again when we consider Macroevolution of populations and ecosystems. Macroevolution of genomes is tightly connected to the evolution of populations: the genome of an organism is just the record of allele replacements in its ancestral lineage. In contrast, Macroevolution of complex phenotypes appears to be mostly independent of Microevolution. Organism Individual ACGATCGACGACGATCGATCGACGATCGA
Topic 15. Lecture 23-24. Macroevolution of Genomes What do we know already about the evolution of sequences? Level-specific generalizations: 1. Sequences a) Mutation strongly affects sequence evolution, and selfish segments are common b) Functionally important segments and sites of genomes usually evolve slower c) Complex organisms have larger genomes, mostly due to noncoding sequences Generalizations concerned with adaptation and complexity: 1. Genetical aspects of adaptive evolution a) Evolution of both coding and non-coding sequences is important for adaptation b) The target for strong positive selection is narrow at each moment c) Tightly related genes can perform rather different functions 3. Origin of novelties a) New non-coding regulatory sites, but not new genes, often appear from scratch
So, what do we want to know, on top of these generalizations and their simple explanations? It makes sense to think of two aspects of sequence evolution. On the one hand, there are properties of sequence evolution that are mostly dictated by selection that acts at the upper levels of organization. We will not consider them here. On the other hand, there are properties of sequence evolution that are not dictated by fitness landscapes in the spaces of molecules, cells, or multicellular organisms. MATEGDKLLGGRFVGSTDPIMEILSSSISTEQRLTEVDIQASMAYAKALEKASILTKTEL... MA+EGDKL GGRF GSTDPIME+L+SSI+ +QRL+EVDIQ SMAYAKALEKA ILTKTEL... MASEGDKLWGGRFSGSTDPIMEMLNSSIACDQRLSEVDIQGSMAYAKALEKAGILTKTEL... Similarity of delta-crystalline sequence (top) to argininosuccinate lyase sequence (bottom), is a sequence-level, and not a molecular-level, phenomenon. Still, before we can do this, I wish to briefly address two fundamental concepts of the theory of sequence evolution that are not directly concerned with any deep understanding of evolution, but are necessary to reconstruct its past course.
Reconstructing the course of past Macroevolution of genomes: Evolutionary distance. Evolutionary distance (ED) between two sequences that diverged from the same ancestral sequence is the number of accepted nucleotide replacements per site. If two sequences can be aligned without gaps (simply placed one above the other), their alignment will contain the fraction 1-M of matches and the fraction M of mismatches. ACACGACACGATGCATACTA |||||| ||||||||| ||| ACACGATACGATGCATGCTA If two sequences are very similar to each other, their ED probably equals to M. However, multiple events per site become important if we consider more dissimilar sequences. Indeed, homoplasy can create a match at a site where multiple substitutions occurred after divergence. Can we estimate the total number of replacements, including hidden ones, from the observed dissimilarity M?
We observe the fraction of mismatches M, but we want to know ED, the total number of replacements that occurred per site. If we know how evolution occurred, we can derive the function that relates M (observable) to ED (unobservable). Then, we invert this function, and estimate unobservable from observable.
In the simplest case, known as 1-parameter Jukes-Cantor model, we assume that all 10 possible nucleotide substitutions (A -> T, A -> G, ...) are equally frequent. If the total substitution rate per site is a, the rate at which matches become mismatches is 2a (any replacement in either sequence will turn a match into a mismatch), and the rate at which mismatches become matches is 2a/3 (only one replacement out of 3 possible ones will turn a mismatch into a match). Thus, This equation can be easily integrated: so that Because, ED=2at, our goal has already been achieved: We can also recover, from the same equation, M as a function of time:
Reconstructing the course of past Macroevolution of genomes: Sequence alignment. Common ancestry of individual nucleotides. If divergence of sequences involved insertions and deletions, nucleotides derived from the same ancestral nucleotide can become shifted. Thus, establishing common ancestry of individual nucleotides from different species requires sequence alignment. Let us consider alignment of just 2 sequences, each of length n. They can be aligned, under reasonable assumptions, in time that is proportional only to n2. How could this be done? One option is to construct a "dot-matrix" that describes matches/mismatches between all the nucleotides in two sequences (hence n2). After this, the best path in this matrix can be found, and this path corresponds to the best alignment. A x x X G x x X T x X T x x A x X x C x X GX x x CX x AX x x A C G T C A G T G A A C G C A T T G A | | | | | | | | A C G T C A G T G A Tricks can be used to find alignments faster, but the basic idea is to consider a dot-matrix.
Reconstructing the course of past Macroevolution of genomes: Orthologous segments. In the field of sequence evolution, homology traditionally means common ancestry. It is necessary to distinguish two kinds of common ancestry ("homology") of sequences - orthology and paralogy. Two segments of different genomes are orthologous if they originated from the same segment of the genome of the last common ancestor. Two segments of the same genome are paralogous if they originated from the same segment, by duplication. Two segments of different genomes are paralogous if they originated from different paralogous segments of the genome of the last common ancestor. The last common ancestor of two modern species, A and B, had two paralogs in its genome (red and purple). Red segments of A an B, originated from ancestral red segment, are each other's orthologs. The same is true for purple segments, of course. Red segment of A is a paralog to purple segments of A and B. Purple segment of A is a paralog to purple segments of A and B.
Orthology is established using the bidirectional best hit test. If for segment a in genome A segment b in genome B provides the best hit when a is compared against the whole genome B, and if a provides the best hit for b, when b is compared against the whole genome A, we conclude that a and b are orthologs. If Nature conspired against us, bidirectional best hit approach may falsely conclude that paralogs are orthologs. Thus, genomic contexts can also be used, when A and B are not too distant. A genome may contain two (or more) orthologs to a segment in some other genome, due to post-divergence duplication.
Now, we are ready for theory of Macroevolution at the sequence level. There are useful partial theories, describing a variety of phenomena: 1) genes and other functional genome segments often form families of paralogs. 2) TEs and other junk genome segments often form families of paralogs. 3) non-recombining sex chromosomes and organelle genomes often undergo profound degeneration. 4) Nucleotide composition (GC-content) often varies greatly along the genome. 5) Genome sizes of even not-too-distant species can differ greatly. 6) at functional nucleotide sites, the strength of selection is often s ~1/Ne. So, let us try to understand these 3 sequence-level Macroevolutionary phenomena:
1) genes and other functional genome segments often form families of paralogs. First, let us review the facts. For example, human genome contains 1434 multigene families of three or more paralogous genes. Some paralogs form clusters and are located close to each other, but many other paralogs are scattered across the genome. A sample of clusters of human paralogous genes, formed by recent duplications.
A majority of genes within a multigene family have at least one very close paralog. KS was estimated for each human gene and its most closely related human paralog.
Now, what do we need to understand? Three things: 1) Why some gene duplications are maintained, and not eliminated by negative selection? 2) What happens to the paralogs, after a duplication is fixed? They can either: i) evolve different functions (neofunctionalization) or ii) each retain only a part of the original function (subfunctionalization). 3) What processes affect the overall properties of multigene families? The "life history" of a successful gene duplication consists of 3 phases: i) its origin by a unique mutation, ii) its fixation within the population, and iii) divergence of paralogs.
Mutations that involve a duplication of a long sequence occur occasionally. A small fraction of duplications that become successfully fixed are probably favored by positive selection. Haploinsufficient genes, such that heterozygotes carrying a loss-of-function allele have low fitness, have more paralogs than haplosufficient genes. If a gene is haploinsufficient, duplicating it may be a good idea!
After a duplication becomes fixed, two things can happen. One of the two paralogs can be lost, reversing the duplication. However, if both paralogs are retained, they will diverge. There are 2 possibilities: subfunctionalization or neofunctionalization. Only in the second case the outcome of a duplication is better than the initial state. subfunctionalization neofunctionalization How to explain the distribution of sizes of families and the excess of similar paralogs? One possibility is episodes of expansion and contraction of a multigene family. There are little data for this scenario. However, paralogs often "talk to each other" through gene conversion, which can explain the apparent excess of "recent" duplications. So, we at least know what questions to ask regarding the evolution of multigene families.
2) TEs and other junk genome segments often form families of paralogs. First, let us review the facts.We already know them: 1. In many species, families of paralogous transposable elements (TEs) constitute a large fraction of the genome. 2. Evolutionary distances between paralogs within a family indicate the time when the family has been formed. 3. In some species (Drosophila) individual TEs are rare, while in others (Mammals) they are mostly fixed. We need to understand factors that control the dynamics of the families of TEs. The ability of TEs to cause their own duplications (transpositions) is the cause of the formation of TE families. But what regulates the number of TEs in a family?
Is there an equilibrium number of TEs within a family? Theoretically, both yes and no answers are possible. Paralogous TEs may help each other to propagate. Thus, an insertion rate grows with the size of a TE family. 1. Equilibrium: insertion rate does not depend on the TE number, elimination rate increases. 2. Equilibrium: both rates increase, but elimination rate increases faster. 3. No equilibrium: both rates increase, but elimination rate increases slower. Unlimited expansion of TEs of a particular kind in the genome must eventually lead to extinction of the host lineage. If so, why did not TEs kill all life?
Another way to ask this question is: what increases the rate of elimination of TEs when their number grows? Apparently, the only force which can eliminate TEs is selection against those host genotypes that carry many of them. Still, there are two options: 1) Selection against genotypes with many TEs may be stronger, due to epistasis. 2) When TEs accumulate, the probability of ectopic recombination increases. Perhaps both these effects are responsible for preventing unlimited expansion of TEs and saving live from extinction.
3) non-recombining sex chromosomes and organelle genomes often undergo profound degeneration. First, let us review the facts. In many clades, sex chromosomes evolved independently. Often, the chromosome restricted to the heterogametic sex (Y or W) never undergoes recombination. Such non-recombining sex chromosomes have only a small number of functional genes, contain a lot of repetitive junk DNA, and encode proteins that carry multiple mildly deleterious amino acid replacements. If males are heterogametic, females are XX, and males are XY. If females are heterogametic, females are ZW, and males are ZZ. Evolutionary degeneration of a non-recombining sex chromosome. Why does it happen? Apparently, four processes contribute to this effect.
Models (a–c) assume that purifying selection against deleterious mutations is less efficient on the Y, and model (d) assumes the same about positive selection for beneficial mutations. (a) Accumulation of weakly deleterious mutations by background selection. (b) Muller's ratchet. (c) Genetic hitchhiking by favorable mutations. (d) Lack of adaptation on the non-recombining Y chromosome.
In fact, long-term degeneration of non-recombining Y chromosomes is not the whole story. Y chromosomes reside only in males, and X chromosomes reside in females 2/3 of the time. Thus, genes with a net male benefit can accumulate on Y chromosome. In contrast, X chromosome can accumulate genes with female benefits. The accumulation of sexually antagonistic alleles on X and Y selects for the suppression of recombination between the nascent sex chromosomes, creating a male-specific region on the Y (MSY). The lack of recombination within the MSY causes genes in this region to degenerate, whereas their homologs on the X might evolve dosage compensation. Next slide shows a more realistic scenario of the evolution of sex chromosomes. A number of open questions remain, but the key process of degeneration of a non-recombining of sex chromosome appears to be well-understood.
Concluding remarks on the evolution of genomes: A genome is a chronicle of past allele replacements, and Macroevolution of genomes can be to a large extent explained through Microevolution of populations. This is good news. The most interesting facets of the evolution of genomes are concerned with their suboptimality - due to mutation-imposed limits on adaptive evolution (responsible for the origin of multigene families), mutational pressures (responsible for proliferation of TEs), and inefficient selection (responsible for degeneration of non-recombining chromosomes). Is accumulation of mildly deleterious junk DNA essential for adaptive evolution? Functional sequences often evolve from junk DNA. However, it is not clear whether availability of junk was ever a limiting factor for adaptive evolution. If yes, efficient selection against junk DNA in unicellular organisms with large populations may prevent evolution of complexity. Are we complex because our ancestors somehow accumulated a lot of junk DNA? OR Do we carry a lot of junk DNA because we are complex and, thus, large? Currently, we do not know the answer.
Quiz So, we know that complex multicellular organisms have large, "bloated" genomes that contain a lot of long introns, transposable elements, and other mostly junk DNA. Two scenarios can be responsible for this correlation: 1) (Complexity as the cause of large genomes). Complex multicellular organisms are physically large. Thus, their populations are necessarily small - and in small populations weak selection against new pieces of junk DNA is inefficient. Thus, genomes became bloated. 2) (Large genomes as the cause of complexity). Initially, the genomes of simple unicellular ancestors of modern complex organisms became bloated - perhaps, these ancestors had low population size due to some reason. After this, complexity and multicellularity evolved, due to recruitment of some initially junk sequences for regulation of gene expression. What kinds of data and analyses could determine, which of the two scenarios correspond to reality?