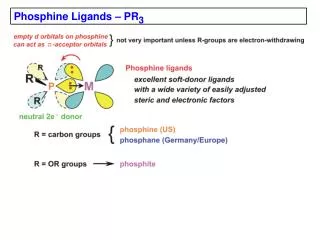
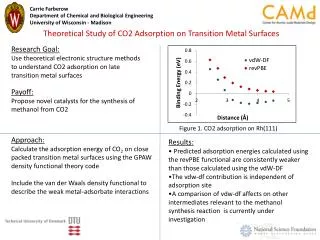
Download

1 / 21
220 likes | 410 Vues
Theoretical study of Scorpionate LigandS. Margarita Mayoral Villa Dec. 2008. Objectives. Examine the properties of the metal- ligand bond in the scorpionate compounds. Examine the variations that could be possible if the metal ion changes its oxidation state.

E N D
Theoretical study of ScorpionateLigandS Margarita Mayoral Villa Dec. 2008
Objectives • Examine the properties of the metal-ligand bond in the scorpionate compounds. • Examine the variations that could be possible if the metal ion changes its oxidation state.
Molecular Physics Methods and Quantum Mechanics • Explains how the electrons have both particles-like and waves-like behaviour. • Use the Schrödinger equation to obtain the energy and other characteristics of the atoms or molecules.
Schrödinger Equation Where: Or in a Hamiltonian form: Where:
Molecular Physics Methods • Hartree-Fock Method (ab initio) • Semiempirical Methods • Density Functional Methods (FDT)
Molecular Orbital Theory (MO) 1) Stationary wave function 2)Born -Oppenheimer approximation: Electronic wave function in a static nuclei field. Where: Is the electronic Hamiltonian Is the electronic wave function Is the effective nuclear potential function 3) Linear combination of atomic orbitals (LCAO)
Self Consistent Method of Hartree-Fock • Is the base of all other methods. • It`s applied in atoms with many electrons, molecules and solids. • It solve the Schrödinger equation using a linear combination of atomic orbitals (LCAO) like the wave function. • It is an iterative method. • When the method finds the minimal energy then that wave function is considered like the correct one, and then the observables are calculated.
Semi-empirical Methods • Use parameters derived of experimental data to simplify the approximation to the Schrödinger equation. • Relatively inexpensive. Appropriated for: • Very large systems. • As a first step in a very large systems. • For ground state molecular systems for which the semi-empirical method is well-parametrized and well-calibrated. • To obtain qualitative information.
Density Functional Methods • Derived from the Thomas-Fermi-Dirac model (1920`s). • Slater`s fundamental work (1950`s). Models electron correlation via functionals of the electron density. • Hohenberg-Kohn theorem (1964). Demonstrated the existence of a unique functional which determines the ground state energy and density exactly. Kohn and Sham propound the approximate functionals employed currently by DFT methods: Where:
Case of study:BisPyrazolylborate (Bp) TrisPyrazolylborate (Tp)
Characteristics to analyze: • Geometry • Homo –Lumo properties • Frequencies • Spectra
Methodology • All the calculations were performed in the Gaussian 03 program. • We use Gauss View for develop the ligands and for visualize the results. • The Hartree – Fock method doesn`t works well with the geometry optimizations. • Then all the calculations were made with Density Functional Theory.
Results We do first the calculations for the ligand: Charge: 0 Multiplicity: 1 And in the second step we do the calculations for: Charge: +1 Multiplicity: 2 Then we can do the comparisons between the results of both ligands.
Conclusions • The better theoretical method in this work is the DFT method using the hibrid exchange-correlation functional: B3LYP, (Becke`s three-parameter formulation) with the 6-31G(d) basis set. • The computational cost for this method is acceptable. • The comparison between the geometries obtained from the calculations with the experimental are very closely. • At the moment, the geometry obtained for charge +1 is very alike to that obtained for charge 0. • We must to compare the spectrum UV-visible obtained from the calculations with that obtained experimentally to be sure that the calculations goes well. • At the moment we can see that the electronic distribution is over the central metal atom in the case of charge 0, and for charge +1 we can see that this is distribuited over the central atom in the case of the HOMO alpha, but for the HOMO beta, this charge seems to go out of the center.
Next work • We must to calculate the Spectrum UV-Vis of the molecule with charge 0 to compare with the experimental Spectrum. • We have to start with the geometry optimization of the Tpligand, using the same method and compare with the experimental one. • Then, confirm the stability, calculate Homo-Lumo, frequencies, spectrum UV-Vis. • Do the same with the charge +1.