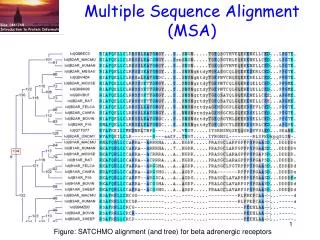
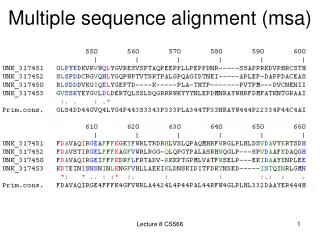
Multiple sequence alignment MSA
Multiple sequence alignment MSA. What is MSA. Comparison of many (i.e., >2) sequences local or global. Why MSA. Biological sequences often occur in families. Homologous sequences often retain similar structures and functions. related genes within an organism


Multiple sequence alignment MSA
E N D
Presentation Transcript
What is MSA • Comparison of many (i.e., >2) sequences • local or global
Why MSA • Biological sequences often occur in families. Homologous sequences often retain similar structures and functions. • related genes within an organism • genes in various species • sequences within a population (polymorphic variants) • MSA reveals more biological information than pairwise alignment • two sequences that may not align well to each other can be aligned via their relationship to a third sequence, thereby integrating information in a way not possible using only pairwise alignments
Edgar R.S. et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature2012485(7399):459-64
Edgar R.S. et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature2012485(7399):459-64
Why MSA • Can be a reasonable way to infer gene function • Characterize protein families by identifying shared regions of homology (conserved regions called motifs), such as active sites • Determine the consensus sequence of several aligned sequences • Establish relationships and phylogenies
What is a sequence motif? • A short conserved region in DNA, RNA or protein sequence • simple combinations of secondary structure elements • motif = supersecondary structure • in proteins, structure motifs usually consist of just a few elements; e.g., the 'helix-turn-helix' has just three • Corresponds to a structural or functional feature in proteins • Shared by several sequences, can be generated by MSA
Examples of motifs beta hairpin helix-loop-helix greek key
What is a protein family? • A protein family is a group of evolutionarily-related proteins. • Proteins in a family descend from a common ancestor and typically have similar three-dimensional structures, functions, and significant sequence similarity. • Members of a protein family may range from very similar to quite diverse. • Currently, over 60,000 protein families have been defined, although ambiguity in the definition of protein family leads different researchers to wildly varying numbers
What is a protein family? • the use of protein family is somewhat context dependent • A common usage is that superfamilies (structural homology) contain families (sequence homology) which contain sub-families. • Example: superfamily PA clan • the largest group of proteases with common ancestry as identified by structuralhomology • has far lower sequence conservation than one of the families tit contains, the C04 family
PA clan structure the double-beta barrel motif
Pfam - http://pfam.sanger.ac.uk/ • Database of protein families that includes their annotations and multiple sequence alignments
What is a domain • Families often share domains. Domain is a part of a protein, and is greater than a motif. • Domain is formed by several motifs packed together. • i.e. domain = tertiary structure • Domain - a conserved part of a given protein sequence and structure that can evolve, function, and exist independently of the rest of the protein chain • One domain may appear in a variety of different proteins • Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions
Pyruvate kinase domains http://en.wikipedia.org/wiki/Protein_domains
Sequence logo Conserved: BIG letters with few others in that space Divergent: small letters with many others in that space
Doing MSA • As with aligning a pair of sequences, the difficulty in aligning a group of sequences varies considerably with sequence similarity. • If the amount of sequence variation is minimal, it is quite straightforward to align the sequences, even without the assistance of a computer program. • If the amount of sequence variation is great, it may be very difficult to find an optimal alignment of the sequences because so many combinations of substitutions, insertions, and deletions, each predicting a different alignment, are possible.
Challenges of the MSA • Finding an optimal alignment of more than two sequences that includes matches, mismatches, and gaps, and that takes into account the degree of variation in all of the sequences at the same time poses a very difficult challenge. • A second computational challenge is identifying a reasonable method of obtaining a cumulative score for the substitutions in the column of an MSA. • Finally, the placement and scoring of gaps in the various sequences of an msa presents an additional challenge.
MSA algorithms • As with the pairwise sequence comparisons, there are two types of multiple alignment algorithms • optimal • heuristic
Optimal algorithms • Extension of dynamic programming to multiple sequences • Exhaustive search • Produce best alignment • Computationally expensive • Not feasible for n>10 sequences of length m>200 residues
Heuristic algorithms • Limit the exhaustive search • Attempt to rapidly find a good, but not necessarily optimal alignment • Most popular methods: • progressive methods (ClustalW) • start from the most similar sequences and progressively add new sequences • iterative methods (MUSCLE) • make initial crude alignment, then revise it
Progressive sequence alignment • The most commonly used algorithm, the most commonly used software ClustalW • Popularized by Feng and Doolitle, often referred by these two names. • Permits the rapid alignment of even hundreds of sequences. • Limitation: the final alignment depends on the order in which sequences are joined. • Not guaranteed to provide the most accurate alignments.
ClustalW • http://www.clustal.org • http://www.ebi.ac.uk/Tools/msa/clustalw2/ • EMBOSS – a free open source software analysis package (European Molecular Biology Open Software Suite) - http://emboss.sourceforge.net • Program emma is a ClustalW wrapper • A variety of EMBOSS servers hosting emma are available, e.g. http://embossgui.sourceforge.net/demo/emma.html • ClustalX – a downloadable stand-alone program offering a graphical user interface for editing multiple sequence alignments • http://www.clustal.org/clustal2/
ClustalW – how it works? Three stages
1st stage The global alignment (Needlman-Wunsch) is used to create pairwise alignments of every protein pair. number of pairwise alignments
1st stage • The raw similarity scores are shown. However, for the next step the distance matrix is needed, and not the similarity one. • Similarity scores must be converted into distances. • Won't tell you how, believe me, it is doable.
2nd stage • A guide tree is calculated from the distance matrix. • The tree reflects the relatedness of all the proteins to be multiply aligned Newick format
Guided tree • Guide trees are not true phylogenetic trees. • They are templates used in the third stage of ClustalW to define the order in which sequences are added to a multiple alignment. • A guided tree is estimated from a distance matrix of the sequences you are aligning. • In contrast, a phylogenetic tree almost always includes a model to account for multiple substitutions that commonly occur at the position of aligned residues.
Construction of guided tree • UnweightedPair Group Method with Arithmetic Mean (UPGMA) • A simple hierarchical clustering method • How it works? http://www.southampton.ac.uk/~re1u06/teaching/upgma/ • Neighbor joining • Uses distance method, distance matrix is an input. • The algorithm starts with a completely unresolved tree (its topology is a star network), and iterates over until the tree is completely resolved and all branch lengths are known.
3rd stage • The multiple sequence alignment is created in a series of steps based on the order presented in the guide tree. • First select the two most closely related sequences from the guide tree and create a pairwise alignment.
3rd stage • The next sequence is either added to the pairwise alignment (to generate an aligned group of three sequences, sometimes called a profile) or used in another pairwise alignment. • At some point, profiles are aligned with profiles. • The alignment continues progressively until the root of the tree is reached, and all sequences have been aligned.
Gaps • “once a gap, always a gap” rule • The most closely related pair of sequences is aligned first. • As further sequences are added to the alignment, there are many ways that gaps could be included. • Gaps are often added to first two (closest) sequences. • To change the initial gap choices later on would be to give more weight to distantly related sequences. • To maintain the initial gap choices is to trust that those gaps are most believable.
Iterative approaches • Progressive alignment methods have the inherent limitation that once an error occurs in the alignment process it cannot be corrected, and iterative approaches can overcome this limitation. • Create an initial alignment and then modify it to try to improve it. • e.g. MUSCLE, IterAlign, Praline, MAFFT
MUSCLE • Since its introduction in 2004, the MUSCLE program of Robert Edgar has become popular because of its accuracy and its exceptional speed, especially for multiple sequence alignments involving large number of sequences. • Multiple sequence comparison by log expectation • Three stages
MUSCLE 1. Draft alignment 2. Improved alignment 3. Refinement Edgar, R. C. Nucl. Acids Res. 2004 32:1792-1797; doi:10.1093/nar/gkh340
MUSCLE online https://www.ebi.ac.uk/Tools/msa/muscle/
>neuroglobin 1OJ6A NP_067080.1 [Homo sapiens] -------------MERPEPELIRQSWRAVSRSPLEHGTVLFARLFALEPDLLPLFQYNCR QFSSPEDCLSSPEFLDHIRKVMLVI---DAAVTNVEDLSSLEEYLASLGRKHRAVGVKLS SFSTVGESLLYMLEKCLGPA-FTPATRAAWSQLYGAVVQAMSRGWDGE---- >rice_globin 1D8U rice Non-Symbiotic Plant Hemoglobin NP_001049476.1 [Oryza sativa (japonica cultivar-group)] MALVEDNNAVAVSFSEEQEALVLKSWAILKKDSANIALRFFLKIFEVAPSASQMFSF-LR NSDVP--LEKNPKLKTHAMSVFVMTCEAAAQLRKAGKVTVRDTTLKRLGATHLKYGVGDA HFEVVKFALLDTIKEEVPADMWSPAMKSAWSEAYDHLVAAIKQEMKPAE--- >soybean_globin 1FSL leghemoglobin P02238 LGBA_SOYBN [Glycine max] ----------MVAFTEKQDALVSSSFEAFKANIPQYSVVFYTSILEKAPAAKDLFSF-LA NGVDP----TNPKLTGHAEKLFALVRDSAGQLKASGTVVAD----AALGSVHAQKAVTDP QFVVVKEALLKTIKAAVGDK-WSDELSRAWEVAYDELAAAIKKA-------- >beta_globin 2hhbB NP_000509.1 [Homo sapiens] ----------MVHLTPEEKSAVTALWGKVNVD--EVGGEALGRLLVVYPWTQRFFES-FG DLSTPDAVMGNPKVKAHGKKVLGAF---SDGLAHLDNLKGTFATLSELHCDKLH--VDPE NFRLLGNVLVCVLAHHFGKE-FTPPVQAAYQKVVAGVANALAHKYH------ >myoglobin 2MM1 NP_005359.1 [Homo sapiens] -----------MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDK-FK HLKSEDEMKASEDLKKHGATVLTAL---GGILKKKGHHEAEIKPLAQSHATKHK--IPVK YLEFISECIIQVLQSKHPGD-FGADAQGAMNKALELFRKDMASNYKELGFQG
MUSCLE vs. Clustal Q = fraction of correctly aligned residues (pairwise) TC = fraction of correctly aligned columns
Logo visualization of the alignment • Make a logo from your alignment • Can be easier to compare • Nice graphic • Students love ‘em • http://weblogo.berkeley.edu/logo.cgi