Comparative Analysis of BCL-2 Proteins in Humans and Mice
Explore the alignments and functional predictions of BCL-2 proteins in humans and mice, highlighting similarities and differences using multiple alignment techniques and correlated mutations.

Comparative Analysis of BCL-2 Proteins in Humans and Mice
E N D
Presentation Transcript
Manual alignment • Difficult for remote homologies • Slow • Low reproductibility • Allows to consider information not included in the sequence
“Equivalent” Aminoácids • Hydrophobic • Ala (A), Val (V), Met (M), Leu (L), Ile (I), Phe (F), Trp (W), Tyr (Y) • Small • Gly (G), Ala (A), Ser (S) • Polar • Ser (S), Thr (T), Asn (N), Gln (Q), Tyr (Y) • Polar and charged are equivalent on the surface • Charged • Asp (D), Glu (E) / Lys (K), Arg (R) • Hard to replace (special function) • Gly (G), Pro (P), Cys (C), His (H)
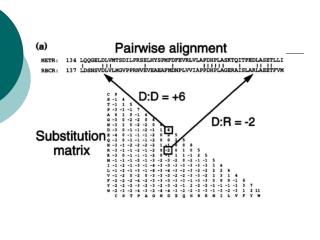
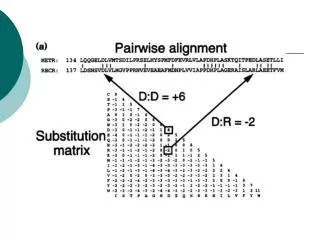
BCL2 Human vs BCL2 Mouse >sp|P10417|BCL2_MOUSE Apoptosis regulator Bcl-2 Length=236 Score = 429 bits (1103), Expect = 6e-119, Method: Composition-based stats. Identities = 214/239 (89%), Positives = 217/239 (90%), Gaps = 3/239 (1%) MAHAGRTGYDNREIVMKYIHYKLSQRGYEWDAGDVGAAPPGAAPAPGIFSSQPGHTPHPA 60 MA AGRTGYDNREIVMKYIHYKLSQRGYEWDAGD AAP GAAP PGIFS QP P PA MAQAGRTGYDNREIVMKYIHYKLSQRGYEWDAGDADAAPLGAAPTPGIFSFQPESNPMPA 60 ASRDPVARTSPLQTPAAPGAAAGPALSPVPPVVHLTLRQAGDDFSRRYRRDFAEMSSQLH 120 R+ ARTSPL+ A AGPALSPVPP VHLTLR+AGDDFSRRYRRDFAEMSSQLH VHREMAARTSPLRPLVA---TAGPALSPVPPCVHLTLRRAGDDFSRRYRRDFAEMSSQLH 117 LTPFTARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEY 180 LTPFTARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEY LTPFTARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEY 177 LNRHLHTWIQDNGGWDAFVELYGPSMRPLFDFSWLSLKTLLSLALVGACITLGAYLGHK 239 LNRHLHTWIQDNGGWDAFVELYGPSMRPLFDFSWLSLKTLLSLALVGACITLGAYLGHK LNRHLHTWIQDNGGWDAFVELYGPSMRPLFDFSWLSLKTLLSLALVGACITLGAYLGHK 236 BH4 BH3 BH1 PhosphoSer
BCL-2 vs BCL-X >emb|CAA57886.1| bcl-x [Rattus norvegicus] Length=233 Score = 172 bits (435), Expect = 2e-41, Method: Composition-based stats. Identities = 93/199 (46%), Positives = 116/199 (58%), Gaps = 13/199 (6%) NREIVMKYIHYKLSQRGYEW----DAGDVGAAPPGAAPAPGIFSSQPGHTPHPAASRDPV 66 N+E+V+ ++ YKLSQ+GY W D + P S P + P NQELVVDFLSYKLSQKGYSWSQFSDVEENRTEAPEETEPERETPSAINGNPSWHLADSPA 64 ARTSPLQTPAAPGAAAGPALSPVPPV--VHLTLRQAGDDFSRRYRRDFAEMSSQLHLTPF 124 A G ++ V P+ V LR+AGD+F RYRR F++++SQLH+TP VN-------GATGHSSSLDAREVIPMAAVKQALREAGDEFELRYRRAFSDLTSQLHITPG 117 TARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEYLNRH 184 TA F VV ELFRDGVNWGRIVAFF FGG +CVESV++EM LV IA WM YLN H TAYQSFEQVVNELFRDGVNWGRIVAFFSFGGALCVESVDKEMQVLVSRIASWMATYLNDH 177 LHTWIQDNGGWDAFVELYG 203 L WIQ+NGGWD FV+LYG LEPWIQENGGWDTFVDLYG 196
BCL-2 vs BCL-X >emb|CAA57886.1| bcl-x [Rattus norvegicus] Length=233 Score = 172 bits (435), Expect = 2e-41, Method: Composition-based stats. Identities = 93/199 (46%), Positives = 116/199 (58%), Gaps = 13/199 (6%) NREIVMKYIHYKLSQRGYEW----DAGDVGAAPPGAAPAPGIFSSQPGHTPHPAASRDPV 66 N+E+V+ ++ YKLSQ+GY W D + P S P + P NQELVVDFLSYKLSQKGYSWSQFSDVEENRTEAPEETEPERETPSAINGNPSWHLADSPA 64 ARTSPLQTPAAPGAAAGPALSPVPPV--VHLTLRQAGDDFSRRYRRDFAEMSSQLHLTPF 124 A G ++ V P+ V LR+AGD+F RYRR F++++SQLH+TP VN-------GATGHSSSLDAREVIPMAAVKQALREAGDEFELRYRRAFSDLTSQLHITPG 117 TARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEYLNRH 184 TA F VV ELFRDGVNWGRIVAFF FGG +CVESV++EM LV IA WM YLN H TAYQSFEQVVNELFRDGVNWGRIVAFFSFGGALCVESVDKEMQVLVSRIASWMATYLNDH 177 LHTWIQDNGGWDAFVELYG 203 L WIQ+NGGWD FV+LYG LEPWIQENGGWDTFVDLYG 196 BH4 BH3 BH1 PhosphoSer
Multiple alignment & functional prediction Aim: Predict function from sequence
General strategies • Group sequences in a “family” (BLAST, PFAM) • Recognize sequence fragments (short) related to functional or structural features
Multiple alignment • Global alignment, more than two sequences • Allow to include the importance of the position in the alignment • Allow to define conserved residues • Residues with functional or structural importance • Tree determinants • Correlated mutations
Multiple alignments • Low similarity, only two sequences: AVTTGLNMWTTAKRPGMDDFYTILLPGLMNCIGLFTAIDMHFFGRKPACEEYFTLVVDGLCNCI • Low similarity, multiple sequences: GIFTDIDMHFYVKKPGLDEFFTLVLRTLCMAAALTTGIDMWTTAKRPDMDDYYTIIIPGLMNCIAVTTGLNMWTTAKRPGMDDFYTILLPGLMNCIGVTTGLNMYFTARRPGLDEFYTLVLRTLCMCL GIFTDIDMHFYVKKPGLDEFFTLVLRTLCMAAAVTTGLNMWTTAKRPGMDDFYTILLPGLMNCIGLFTALNMHFFGRKPACEEYFTLVVDGLCNCI
Tree determinants • Define subfamilies • Relevant to philogeny S1 GIFTDIDMHFYVKKPGLDEFFTLVLRTLCMAAS2 ALTTGIDMWTTAKRPDMDDYYTIIIPGLMNCIS3 AVTTGLNMWTTAKRPGMDDFYTILLPGLMNCIS4 GVTTGLNLYFTARRP--DEFYS-VLRTLCMCL S5 GIFTDIDLHFYVKKP--DEFFSLVLRTLCMAAS6 AVTTGLNLWTTAKRP--DDFYSILLPGLMNCIS7 GLFTALNLHFFGRKP--EEYFSLVVDGLCNCI
Correlated mutations • Concerted changes in two or more conserved positions • Reveal positions of structural interaction GIFTDIDMHFYVKKPGL DEFFTLVLRTLCMAAALTTGIDMWTTAKRPDM DDYYTIIIRGLMNCIAVTTGLDMWTTAKRPGM DDFYTILLRGLMNCIGVTTGLDMYFTARRPGL DEFYTLVLKTLCMCL GIFTDIRMHFYVKKPGL DEFFTLVLDTLCMAAAVTTGLRMWTTAKRPGM DDFYTILLDGLMNCIGLFTALRMHFFGRKPAC EEYFTLVVEGLCNCI R-D D-R
Software • ClustalW • Makes global pairwise alignments building “clusters” of similar sequences • Tcoffee • Slower than clustalw but more precise for low similarity • Combines global/local alignments
Profiles • Also known as Position-specific score matrix (PSSM). • Give scores for amino acids or gaps specific to sequence positions • Quantitative approach to include the role of positions
F K L L S H C L L V F K A F G Q T M F Q Y P I V G Q E L L G F P V V K E A I L K F K V L A A V I A D L E F I S E CI I Q F K L L G N V L V C A 0 01 0111 0 1 0 C 0 0 0 0 0 02 0 01 D 0 0 0 0 0 00 0 01 E 0 1 0 0 021 0 0 0 F 5 0 0 1 0 0 0 0 1 0 G 0 0 0 03 0 0 0 01 H 0 0 0 0 01 0 0 0 0 I 0 011 0 0 031 0 K 0 4 0 01 0 0 0 01 L 1 023 0 0 033 0 M 0 0 0 0 0 0 01 0 0 N 0 0 0 0 01 0 0 0 0 P 0 2 0 0 0 0 0 0 0 0 Q 0 0 0 0 02 0 0 02 R 0 0 0 0 0 0 0 0 0 0 S 0 0 0 02 0 0 0 0 0 T 0 0 0 0 0 01 0 0 0 V 0 022 0 02 0 11 W 0 0 0 0 0 0 0 0 0 0 Y 1 0 0 0 0 0 0 0 0 0
Profiles, simplest calculation Frecuency of i at position j Nij/NS Mij = log fi Standard frequency of i. > 0: Position j is rich in aa. i Mij 0: Normal < 0: Position j is poor in the aa. i
Profiles, improvements • Include amino acid frequency according to protein family • Include also classical similarity matrices • Allows equivalent amino acids that do no appear in the available alignment.
F K L L S H C L L V F K A F G Q T M F Q Y P I V G Q E L L G F P V V K E A I L K F K V L A A V I A D L E F I S E CI I Q F K L L G N V L V C A -18 –10 -1 -8 8 -3 3 -10 -2 -8 C -22 -33 -18 -18 -22 -26 22 -24 -19 -7 D -35 0 -32 -33 -7 6 -17 -34 -31 0 E -27 15 -25 -26 -9 23 -9 -24 -23 -1 F 60 -30 12 14 -26 -29 -15 4 12 -29 G -30 -20 -28 -32 28 -14 -23 -33 -27 -5 H -13 -12 -25 -25 -16 14 -22 -22 -23 -10 I 3 -27 21 25 -29 -23 -8 33 19 -23 K -26 25 -25 -27 -6 4 -15 -27 -26 0 L 14 -28 19 27 -27 -20 -9 33 26 -21 M 3 -15 10 14 -17 -10 -9 25 12 -11 N -22 -6 -24 -27 1 8 -15 -24 -24 -4 P -30 24 -26 -28 -14 -10 -22 -24 -26 -18 Q -32 5 -25 -26 -9 24 -16 -17 -23 7 R -18 9 -22 -22 -10 0 -18 -23 -22 -4 S -22 -8 -16 -21 11 2 -1 -24 -19 -4 T -10 -10 -6 -7 -5 -8 2 -10 -7 -11 V 0 -25 22 25 -19 -26 6 19 16 -16 W 9 -25 -18 -19 -25 -27 -34 -20 -17 -28 Y 34 -18 -1 1 -23 -12 -19 0 0 -18
F K L L S H C L L V F K A F G Q T M F Q Y P I V G Q E L L G F P V V K E A I L K F K V L A A V I A D L E F I S E CI I Q F K L L G N V L V C A -18 –10 -1 -8 8 -3 3 -10 -2 -8 C -22 -33 -18 -18 -22 -26 22 -24 -19 -7 D -35 0 -32 -33 -7 6 -17 -34 -31 0 E -27 15 -25 -26 -9 23 -9 -24 -23 -1 F 60 -30 12 14 -26 -29 -15 4 12 -29 G -30 -20 -28 -32 28 -14 -23 -33 -27 -5 H -13 -12 -25 -25 -16 14 -22 -22 -23 -10 I 3 -27 21 25 -29 -23 -8 33 19 -23 K -26 25 -25 -27 -6 4 -15 -27 -26 0 L 14 -28 19 27 -27 -20 -9 33 26 -21 M 3 -15 10 14 -17 -10 -9 25 12 -11 N -22 -6 -24 -27 1 8 -15 -24 -24 -4 P -30 24 -26 -28 -14 -10 -22 -24 -26 -18 Q -32 5 -25 -26 -9 24 -16 -17 -23 7 R -18 9 -22 -22 -10 0 -18 -23 -22 -4 S -22 -8 -16 -21 11 2 -1 -24 -19 -4 T -10 -10 -6 -7 -5 -8 2 -10 -7 -11 V 0 -25 22 25 -19 -26 6 19 16 -16 W 9 -25 -18 -19 -25 -27 -34 -20 -17 -28 Y 34 -18 -1 1 -23 -12 -19 0 0 -18
F K L L S H C L L V F K A F G Q T M F Q Y P I V G Q E L L G F P V V K E A I L K F K V L A A V I A D L E F I S E CI I Q F K L L G N V L V C A -18 –10 -1 -8 8 -3 3 -10 -2 -8 C -22 -33 -18 -18 -22 -26 22 -24 -19 -7 D -35 0 -32 -33 -7 6 -17 -34 -31 0 E -27 15 -25 -26 -9 23 -9 -24 -23 -1 F 60 -30 12 14 -26 -29 -15 4 12 -29 G -30 -20 -28 -32 28 -14 -23 -33 -27 -5 H -13 -12 -25 -25 -16 14 -22 -22 -23 -10 I 3 -27 21 25 -29 -23 -8 33 19 -23 K -26 25 -25 -27 -6 4 -15 -27 -26 0 L 14 -28 19 27 -27 -20 -9 33 26 -21 M 3 -15 10 14 -17 -10 -9 25 12 -11 N -22 -6 -24 -27 1 8 -15 -24 -24 -4 P -30 24 -26 -28 -14 -10 -22 -24 -26 -18 Q -32 5 -25 -26 -9 24 -16 -17 -23 7 R -18 9 -22 -22 -10 0 -18 -23 -22 -4 S -22 -8 -16 -21 11 2 -1 -24 -19 -4 T -10 -10 -6 -7 -5 -8 2 -10 -7 -11 V 0 -25 22 25 -19 -26 6 19 16 -16 W 9 -25 -18 -19 -25 -27 -34 -20 -17 -28 Y 34 -18 -1 1 -23 -12 -19 0 0 -18
Profiles • The use of profiles increases the information available and allow to extract “family” features opposite to individual sequence features
HMM profiles • Statistical models (Hidden Markov Models) to build profiles. • The model is “trained” using multiple alignments to determine evolution probabilities. • They contain a “theoretical machinery” that allows to understand sequence relatioships in a quantitative basis.
The twilight zone • Identity below 25 % • Structural similarity exists but it is difficult to identify it using standard methods
PSI-BLAST • Blast search based on profiles prepared dynamically: • Standard Blast search • Building of a position-specific score matrix (PSSM) from the alignment • New search against the profile • Repeat until self-consistence
PSI-BLAST • PSI-BLAST cannot find what Blast cannot detect • E-value for inclusion/exclusion must be chosen carefully (0.01) • Some times the system goes to non-sense results • Number of iterations is usually kept small
Motifs • Motif: short sequence fragment. Highly conserved, related to some structural or functional feature • Conserved in distant homologues, due to functional or structural restrictions • Help to functional prediction and to detect remote homology
X(12) H C X(3,5) X(2,4) H C C-x(2,4)-C-x(12)-H-x(3,5)-H
Motifs • How to express motifs? • Regular expressions, patterns • Profiles (PSSM) • HMMs (Hidden Markov Models) (PFAM)
Regular expressions ALRDFATHDDF SMTAEATHDSI ECDQAATHEAS A-T-H-[DE]
Regular expressions Never E o D 1 aa [AC]-x-V-x(4)-{ED} A o C 4 aa Sólo V
Regular expressions one or none <A-x-[ST](2)-x(0,1)-V Two S o T N-Terminal
Building regular expressions • From multiple alignments • Manual • Pattern building software • eMotif, PRATT, Teiresias,…
Pattern databases • PROSITE • Known sequence motifs • Active site signatures • Interaction sites • Modification sites (glycosilation, phosphorilation, ...)
[GA]-x(1,2)-[DE]-x-Y-x-[STAP]-x-C-[NKR]-x-[CH]-[LIVMFYWH] G GQ D L Y V P V C R L C Y