6. Gas chromatography
6. Gas chromatography. Adv. Chrom . Revision. 1. How does a gas chromatograph separate a mixture? attraction to the column vs evaporation 2. What limitation on analytes does gas chromatography have? must be volatile


6. Gas chromatography
E N D
Presentation Transcript
6. Gas chromatography Adv. Chrom.
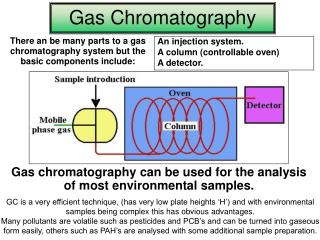
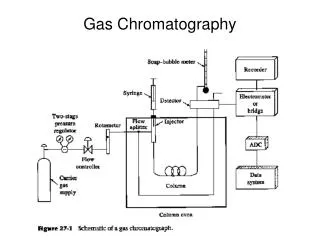
Revision 1. How does a gas chromatograph separate a mixture? • attraction to the column vs evaporation 2. What limitation on analytes does gas chromatography have? • must be volatile 3. Draw a block diagram showing the essential components of a typical gas chromatograph. Oven Carrier gas Injection port Column Detector Readout
Revision 4. What carrier gases are commonly used in gas chromatography? What component of the chromatography system is the carrier gas? • N2, He, mobile phase 5. What is the effect on elution of increasing the temperature? • decreases the time in the column (retention time) 6. How is the stationary phase chosen for a given analysis, e.g. ethanol in alcoholic beverages? • similar polarity to analytes
Revision 7. Why is the injection port heated? • to ensure the sample is vaporised immediately 8. What are the two common methods for determining the identity of compounds by GC? • retention time matching, spiking
Columns • packed column in three different components: • the tubing itself, • the powder or packing • the liquid stationary phase
Tubing & packing material • most commonly made from stainless steel • some are glass • stainless steel columns can be uncoiled and emptied of contaminated material, repacked and recoiled • glass columns - packing must be blown out and in with compressed air • the powder itself is not the stationary phase • an inert support material onto which the stationary phase is coated • essential properties of the support material • small and uniform particle size • large surface area
Stationary phases • a liquid coated in thin layers on the support in packed columns or the walls in capillary columns • various requirements: • low volatility • thermal stability • chemically inert • two main types: • polysiloxanes – non-polar with various substituents, eg methyl, phenyl • polyethylene glycols (PEGs) - polar
Exercise 6.1 What would be a suitable column for the separation of: • (a) separation of petrol components • DB1 • (b) soil extract containing DDT and other pesticides • DB5
Column preparation & conditioning • support is coated with stationary phase • making a solution of the liquid in a volatile solvent • producing a slurry with the appropriate amount of support • evaporating the solvent off • proportion of stationary phase – loading (1-10%w/w) • new column must be conditioned • run at maximum temperature for an extended period with the carrier gas flowing • any remaining solvent or other chemicals involved in manufacture are removed • carrier gas must be flowing whenever the column is heated
Are packed columns a dying breed? • capillary columns separate mixtures more rapidly and with better resolution • so why bother with packed columns? • they can cope with: • high levels of non-volatiles (eg salts, sugars) • higher sample volumes • can be repacked at little cost • once a capillary column stops working, that’s it
Detectors • respond to nanogram or less quantities of analyte passing through the detector within a few seconds • desirable properties for a detector include: • wide linear response • good stability with time • response to all compounds
Thermal conductivity (TCD) • compares the thermal conductivity of the eluant gas with the carrier gas • senses the change in resistance of a current-carrying filament as a result of changes in the composition of the gas flowing over it • an eluting compound changes the thermal conductivity of the gas, and thus the conduction properties to the filament • use a pair of filaments • one placed before the injection port as a reference • the second at the end of the column for the eluant gas • best carrier gas is helium (very different thermal cond.)
Adv. & disadv. • simplicity • cheapness • response to all species • non-destructive • not very sensitive
Flame ionisation (FID) • measures the electrical conductivity of the gas stream from the column • after it passes through a hydrogen/air flame • combustible species produce ions as intermediates in the burning process • ions are collected by a measuring electrode placed over the flame • most sensitive to organic compounds with long C chains • doesn’t respond to non-combustibles, eg water, N2 (ideal carrier gas – cheaper than He)
Adv. & disadv. • very sensitive to hydrocarbons (1 ng/mL) • insensitive to water • relatively cheap ($5-10,000) • require extra gas supplies (hydrogen, air) • can be hard to get lit
Qualitative analysis • RT comparison • quicker • prone to temp. & flow variations • spiking • more reliable • slower
Quantitative analysis • how to measure the instrument response • how to calibrate that response against standards Measuring response • a peak on • a computer screen • a roll of plain or chart paper • truest measure of the amount of compound producing that peak is the area under the peak
Measuring instrument response • electronic integration of peak area • if not available, manual measurement of peak height is better Class Exercise 6.2 • If peak area is the true measure of the amount of compound, why is peak height recommended for measurements on chart paper? • peak are normally very narrow • can’t measure accurately 1-3 mm
Calibration • responses changes between analyte and from day to day • can use normal calibration standards • matrix interference is not a problem in chromatography so std addition not required • the main error in variation in injection volume, especially if done manually • internal standards are used to correct for this • see Chapter 8
Internal standard choice • normal reasons • plus • should not overlap the analyte(s) in the chromatogram • should not extend the run time if possible • these two can be bent slightly • adjust conditions if small overlap occurs • extending run time may be unavoidable, so minimise
Example 6.1 Ethanol to ethanoic acid Possible Int. Stds (retention times) methanol (2) ethanol (2.5) 1-propanol (4) ethanoic acid (4.5) propanoic acid (6.5) Totally unsuitable • ethanoic acid and ethanol – are or may be present Best • Methanol is the quickest, but is quite different to the main analyte, ethanoic acid • 1-propanol fits nicely between the two analytes • Propanoic acid extends the run time • 1-propanol would be best.
Exercise 6.2 Cyclohexanol to cyclohexanone (in hexane) Unsuitable • cyclohexanol, cyclohexanone, hexane – already present Best • cyclopentanone • doesn’t extend run or overlap, similar to product Others 2-hexanone – 2nd choice (peak close to cyclohexanone) 2-hexanol – extends run & similar to minor component
Exercise 6.3 Main products: 2-nitrotoluene and 4-nitrotoluene Minor product: 2,4-dinitrotoluene Unreacted: toluene • should be similar to main products • 3-nitrotoluene is closest, but would be very hard to separate • nitrobenzene still similar (1 CH3 less), lower bp, so shorter RT
Optimisation • the longer a species is retained within the system, the more its peak will spread out: peak broadening Exercise 6.4 Why is peak broadening a problem? • loss of resolution, difficult to measure peak area
Factors affecting elution • the type of column (packed or capillary) • the polarity of the stationary phase • the temperature of the oven • the flow rate of the carrier gas • optimum flow rate (AIT) • temperature main means of improving separation • effect (temp vs time) not linear
Optimum separation • the analyte peak(s) are well-resolved from other peaks • all components are eluted in as short a time as possible • peaks do not have to be completely separated Exercise 6.5 all peaks do not have to be perfectly separated • can measure area/height accurately (<20% overlap; “ankles”) all peaks must be out before the next injection • peaks still in the column will appear in the middle of the next run
Exercise 6.6 (a) analytes A & B • basically OK • A & B could be a little closer • C & D could be sped up all cpds of interest • not OK • C & D overlap too much
Exercise 6.6 (c) analyte is C • definitely not good • C very slow • D hasn’t even appeared
Temperature conditions • same temp. all the way: isothermal • not possible for some samples • need to change (increase) temperature through run: temperature programming • low temp. at start to separate fast cpds • inc. temp. to hurry out slow ones Exercise 6.7 • no broadening for slower peaks 45 isoth. 145 isoth. ?
Measuring separation • by theoretical plate concept • larger N, more efficient • measured from chromatogram • packed columns: 1000-2000 plates/m • capillary: 2000-3000 • main gain in efficiency for capillary columns due to greater length
Exercise 6.8(a) • tr = 4.35 min • tw = 26.3 s • 3 m packed column • convert tr to seconds = 261 (or tw to minutes = 0.422) • 1703 ÷ 3 = 568/m • not great • 6.8(b): 1849/m, OK just
Peak overlap • numerical check • > 1 is fine Exercise 6.9 • remember to convert to same time units • OK