Download
1 / 36
400 likes | 777 Vues
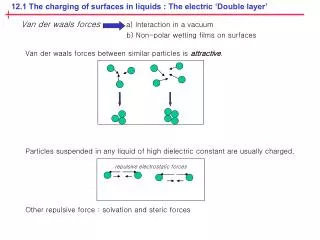
12.1 The charging of surfaces in liquids : The electric ‘Double layer’. repulsive electrostatic forces. Van der waals forces a) Interaction in a vacuum b) Non-polar wetting films on surfaces.

E N D
12.1 The charging of surfaces in liquids : The electric ‘Double layer’ repulsive electrostatic forces Van der waals forces a) Interaction in a vacuum b) Non-polar wetting films on surfaces Van der waals forces between similar particles is attractive. Particles suspended in any liquid of high dielectric constant are usually charged. Other repulsive force : solvation and steric forces
Two charging mechanism of a surface in a liquid 1) By the ionization or dissociation of surface groups -COOH -COO- + H+ 2) By the adsorption of ions from solution onto a previously uncharged surface Ca2+ +H3N-C-COO- surface Fig.12.1 Ions bound to a surface are not rigidly bound but Can exchange with other ions in solution; their lifetime on A surface can be as short as 10-9s or as long as many hours. Stern or Helmholtz layer Electric double layer The final surface charge is electroneutral.
12.2 Charged surfaces in water (no added electrolyte) The counterion distribution and force between two similarly charged planar surfaces in a pure liquid such as water Colloidal particles, clay sheets, surfactant micelles or bilayers whose surfaces contain ionizable groups interact in water H3O+ surface surface Thick films of water build up on an ionizable surface such as glass OH- 12.3 The POISSON-BOLTZMANN (PB) EQUATION = zeψ + kT log ρ(12.1) • : chemical potential of any ion Ψ : electrostatic potential ρ : number of density of ions at any point x between surfaces z : valency of ions at any point x between surfaces, ex) Na+ +1, Cl- -1 Since only differences in potential are ever physically meaningful, Ψ0 = 0 at the midplane (x =0), ρ = ρ0, (dΨ/dx)0 = 0 by symmetry
From the equilibrium requirement, the chemical potential is uniform throughout. Boltzmann distribution of counterions at any point x ρ = ρ0e- zeψ/kT (12.2) Poisson equation for the net excess charge density at x zeρ = -єє0(d2Ψ/dx2) (12.3) POISSON-BOLTZMANN EQUATION d2Ψ/dx2 = -zeρ/єє0 = -(zeρ0/єє0)e- zeψ/kT (12.4) The PB equation gives Ψ, electric field E = ∂Ψ/∂x and ρ at any point x in the gap between the two surfaces. 12.4 Surface charge, electric field and counterion cincentration at a surface Two boundary condition • Symmetry requirement : E0 = (dΨ/dx)0 = 0 • 2) Requirement of overall electroneutrality
i.e. (12.5) Fig 12.2 Two negatively charged surfaces of surface charge density σ separated a distance D in water. Ionic distribution There exists an important general relation between the concentration of counterions a either surface and at the midplane. Differentiating ρ = ρ0e- zeψ/kTand then using d2Ψ/dx2 = (zeρ0/єє0)e- zeψ/kT (12.6)
(12.7) This equation gives ρ at any point in terms of ρ0 at the midplane and (dΨ/dx)2 at x (dΨ/dx)D/2 = (dΨ/dx)s = Es = σ/єє0 (12.8) This result shows that the concentration of counterions at the surface depends only on the surface charge densityσ and the counterion concentration at the midplane 12.5 Counterion concentration profile away from a surface The above equations arethe ionic distributions near planar charged surfaces.
To proceed further for specific case PB Eq. [-zeρ/єє0 = -(zeρ0/єє0)e- zeψ/kT] can be satisfied by (12.9) or (12.10) constant K (12.11) We obtain the electric fields and counterion distribution profile Ψ = 0 and dΨ/dx = 0 at x = 0 differentiating Eq. (12.9) and using (12.12) (12.13)
(PB equation) Monte Carlo simulation Fig. 12.3. Monovalent counterion concentration profile between two charged a distance 2.1nm apart in water. 12.6 Origin of the ionic distribution, electric field, surface potential and pressure • The first thing to notice is that if there no ions between two similarly charged surfaces, • there would be no electric field in the gap between them Charged surface Charged surface E=σ/2εε0 Cancel out to 0
The reason why the counterions build up at each surface is repulsive electrostatic interaction between the counterions and their entropy of mixing which determine ρx, Ψx and Ex between the surfaces. Charged surface Charged surface The combined thickness of the stern and Helmholtz layers δ is of a few angstroms and transiently bound counterions. (12.14) Fig. 12.4. The locations of the cheged surface groups were at the some small distance δ within the surface.
2) The origin of the repulsive force or pressure between the two surfaces is entropic, not electrostatic. Charged surface Charged surface The repulsive osmotic pressure between counterions maintain the diffuse double layer 12.7 The pressure between two charged surfaces in water : the contact value theorem. Insert = zeψ + kT log ρ into (∂P/∂x)x,T = (∂/∂x)x,T (12.15) zeρ = -єє0(d2Ψ/dx2)
(12.16) The above equation gives the pressure P at any point x between the two surfaces At equilibrium, Px(D) should be uniform throughout the gap (12.7) using (12.17a) (12.17b) Px(D) is independent of x and depends only on the increased ionic concentration. We may therefore drop the subscript x from Px(D). Insert (12.8) into the above Using ρs(∞) = ρ0(∞) + σ2/2єє0kT 0 (12.8)
The pressure is also given by the increase in the ion concentration at the surfaces. This important equation is the contact value theorem. Application 1) double-layer interaction 2) solvation interaction (Ch. 13) 3) polymer-associated steric and depletion interaction (Chap. 14, 18) 4) undulation, peristaltic and protrusion forces between fluid membranes (Chap. 18) For specific case, the pressure may also be expressed in terms of K. (12.11) (12.18) At smaller distances the measured forces are more repulsive than expected due to the steric-hydration interactions between the thermally mobile hydrophilic headgroups that characterize these surfaces. (Chap. 14, 18) Fig. 12.5. Measured repulsive pressure between charged bilayer surfaces in water.
Repulsive electrostatic forces also control the long-range swelling of clays in water. water K+ Ca+ Na+ clay In the case of charged spherical paticles in water the long-range electrostatic repulsion between them can result in an ordered lattice of particles even when the distance between them is well in excess of their diameter. (Fig. 10.6g)
12.8 Limitations of The Poisson-Boltzmann Equation ● PB equation Break down !!! at small distances where it no longer describes the ionic distribution, forces between two surfaces (1) Ion-correlation effects the mobile counterions in the diffuse double layer constitute a polarizable layer two apposing conducting layers : attractive van der Waals force ( not included in the PB equation at small distances, more significant (2) Finite ion size (steric) effects enhance the repulsion between two surfaces (3) Image forces additional repulsion (4) Discreteness of surface charges contribute an attractive force (5) Solvation forces short-range solvation or hydration forces : attractive, repulsice or oscillatory (chap 13)
12.9 Thick Wetting Films ● large distances D ( surface charge density () : high ) at any point x : at the surfaces : KD / 2 / 2 ( K / D ) so, P(D) = 2O ( kT / ze )2 D2 (12.20) Langmuir equation : equilibrium thickness of thick wetting films of water on glass surfaces ● disjoining pressure of a film water-air surface - replaces the midplane (d = D / 2 ) so, P(d) =O ( kT / ze )2 / 2d2 (12.21) this repulsive pressure 이것은 surface에 liquid film이 spread되는 현상을 유발 !!! (12.12)
12.9 Thick Wetting Films ● P(d) = -mgH / v = ( kT / v ) log ( P / Psat ) (12.22) H : the height of the film above the surface of the bulk liquid m / v = , P / Psat = relative vapour pressure if water condenses on a charged surface from undersaturated vapour - the film thickness (d) : increase ( H 0, P Psat ) ● application capillary tube에서 water가 예상보다 더 높게 올라간다 : water가 capillary의 inner surface를 젖게 해서 film을 형성함으로써 위와 같은 원인으로 dry radius보다 실제 effective radius는 더욱 적다 !! - 그래서 생각했던 값보다 water가 capillry안에서 더 높게 올라간다
12.10 limit of Small Separation : Charge Regulation ● at small separation, D 0, (12.12)로 부터 K2 - ze / OkTd (12.19) 로 부터 P( D 0 ) = -2 Kt / zeD (12.23) (12.13) K2 = ( ze)2 0 / 2 OkTd (12.11) x = 0 e -ze / kT = 0 / cos2 Kx so, -2 / zeD = number density of counterion in the gap pressure of (12.23) : osmotic pressure P = kT of an ideal gas ● charge regulation P( D 0 ) = -2 Kt / zeD : = constant일 때 …… unrealistic 실제로는 2개의 surface가 molecular contact를 할 정도로 가까워져서 counterion들이 original surface sites에 readsorption을 할 때, D 0 이면 는 charge density 값이므로 constant하지 않고 작아진다 so, constant surface charge라는 가정으로부터 계산된 값보다 effective repulsion의 값이 줄어든다 Counterion density profile x = s = 0 = -2 / zeD at all x (12.24)
12.11 Charged Surfaces In Electrolyte Solutions <Isolated surface> : at surface x=o ● to understand the double-layer interaction between two surfaces at small separation : ionic distribution adjacent to an isolated surface in contact with an electrolyte solution ● the Boltzmann distribution of ions I at x xi = I e -Ziex / kT (12.25) at x = 0 0i = I e -Zieo / kT (12.26) ● if solution containing H+OH- + Na+Cl- + Ca 2+Cl2- [H+]x = [H+] e - ex / kT[H+]o = [H+] e - eo / kT [Na+]x = [Na+] e - ex / kT[Na+]o = [Na+] e - eo / kT [Ca2+]x = [Ca2+] e - 2ex / kT[Ca2+]o = [Ca2+] e - 2eo / kT [Cl-]x = [Cl-] e +ex / kT[Cl-]o = [Cl-] e +eo / kT (12.27)
12.12 The Grahame Equation ● total concentration of ions at an isolated surface of charge density = 0.2 C m-2 at 25oC 2 / 2 O kT = 7.0 1027m-3 = 11.64 M 1:1 electrolyte such as NaCl [Na+]o + [Cl-] o = 11.64 + [Na+] + [Cl-] = 11.64 + 2 [Na+] = 11.64 + [NaCl] (12.29) 2:1 electrolyte such as CaCl2 [Ca2+]o + [Cl-] o = 11.64 + [Cl2+] + [Cl-] = 11.64 + 3 [Ca2+] = 11.64 + [CaCl2] (12.29) (12.8) (12.28)
12.12 The Grahame Equation ● the Grahame equation we can find the relation between the surface charge density and the surface potential o 2 = 2O kT ( ) from (12.26), (12.28) = 2O kT { [Na+]e - eo / kT + [Ca2+] e - 2eo / kT + [Cl-]e +eo / kT - [Na+] - [Ca2+] + [Cl-] } ( [Cl-] = [Na+] + 2[Ca2+] ) 2 = 2O kT { [Na+] (e - eo / kT + e +eo / kT - 2 ) + [Ca+] (e - 2eo / kT + 2e +eo / kT - 3)} Grahame equation = = (12.30) The Grahame equation으로부터 0를 계산할 수 있다 once is known, from the individual ionic concentrations at each surface 0i ( 0i는 (12.26) or (12.27)로 부터 얻을 수 있다
12.13 Surface Change and Potential in The Presence of Monovalent Ions ● at constant surface charge density the surface potential falls progressively as the electrolyte concentration rises surface 에서의 ionic concentration을 결정 !!! in 10-7M 1:1 electrolyte, where 0 = -477.1mV
12.13 Surface Change and Potential in The Presence of Monovalent Ions ● 대부분의 경우에 나 0는 solution의 condition이 변하면서 constant하게 유지되지 않는다 !!! because ionizable surface site가 모두 dissociated 되지 않고 부분적으로 solution으로 부터의 specific ion의 binding에 의해서 neutralized 되기 때문 exchangeable ion inert ion : do not bind to the surface ● if only protons이 negatively charged surface에 binding한다면, Kd (dissociation constant) (12.32) [S-]0 : concetration or surface density of negative surface site (dissociated) [H+]0 : proton configuration at the surface [SH]0 : density of neutral site (12.33) 0 : maximum possible charge density (if all sites were dissociated) : the fraction of sites dissociated ● 이것을 Grahame Equation과 결합하면 (mized 1:1 electrolyte of NaCl + HCl)
12.13 Surface Change and Potential in The Presence of Monovalent Ions ● if Kd is very large (high surface charge, weak binding of proton) = 0=constant fixed surface charge density if Kd takes on a more typical value, the effect can be quite dramatic !! : Kd = 10-4 M, 0 = -0.2 Cm-2 in 0.1M NaCl bulk soution at pH 7 - 0 = -118mV = 0.91 - proton은 surface site의 9%만 neutralize at Ph 5 - 0 = -73mV = 0.36 - 36%의 site가 dissociated되어 남아 있음 0 and will vaty as the salt concentration or pH is changed !! Proton : potential determining ion
12.14 Effect of Divalent Ions ● at constant sruface charge density, relatively small amounts of divalent ions lower the magnitude of 0 0 is determined solely by the devalent cations once their concentration is greater than about 3% of the monovalent ion concentration !!! when the bulk concentration of Ca 2+ is much smaller than that of Na+ the surface may have a much higher local concentration of Ca 2+ 100mM NaCl + 3mM CaCl2 where 0 = -100mV the concentration of Ca 2+ at the sruface [Ca 2+ ] = 310-3 e+200/25.7 =7M [Na+] = 0.1 e +100/25.7=5M ● at high surface concentration divalent ions often bind to negative surface sites : lowering , reducing 0 in the case of trivalent ions such as La 3+ : bulk concentrations in excess of 10-5 M neutralize a negatively charged surface charge reversal !!! In most cases, ion binding tends to lower both and 0 as the concentrations of these ions increase
Potential : proportional to the surface charge density For 1:1 electrolytes (NaCl) For 2:1 and 1:2 electrolytes (CaCl2) For 2:2 electrolytes (MgSO4) 12.15 The Debye Length ● low potential 에서 Grahame equation은 다음과 같이 simplify 되어진다 (12.35) = (12.14) for capacitor : two plates are separated by a distance 1/k charge density ± , potential difference 0 charged capacitor give rise to the name “diffuse electric double layer” describe - ionic atmosphere near a charged surface - thickness : Debye Length 1/k ● Debye Length noly depends on the properties of the liquid (12.35) (12.36) 1/k (12.37)
(12.39) (12.38) (12.41) (12.40) (12.42) 12.16 Variation of Potential and Ionic Concentrations away from a Charged Surface5 The Debye Length ● the potential gradient at any distance x from an isolated surface by (12.27) for 1:1 electrolyte can be integrated using Gouy-Chapman theory !!! - high potentials 1 while for low potentials, Eq.(1239) Debye-Huckel equation the Debye Length 1/k : the characteristic decay length of the potential symmetrical 1:1 electrolytes(NaCl) Debye-Huckel Equation asymmetrical 2:1 and 1:2 electrolytes (CaCl2) Grahame
12.16 Variation of Potential and Ionic Concentrations away from a Charged Surface5 The Debye Length Potential and ionic density profiles 1:1 electrolyte (NaCl) near a surface of charge density = -0.0621Cm-2 calculated form Eqs(12.39)and (12.25) with 0 = -66.2mV from Grahame equation
12.17 The electrostatic double-layer interaction between charged surfaces in electrolyte Electrolyte solution 내의 두 charged surface 간의 interaction pressure를 구하기 위해서 Section12.7에서 식 12.7에서 위의 식에 대입하고 으로 놓으면 P를 midplane의 excess osmotic pressure라고 간단히 나타내면, 1:1 electrolyte 에서…
이 작다고 가정하고 x=D/2에서 단지 potential의 합이라고 가정하면 앞에서 나왔던 식 에 x=D/2를 대입하면 과 같은 식이 나오고 이를 앞의 식에 대입하면 Repulsive pressure between two planar • Weak overlap approximation • never exceed unity ● At low surface potential(약 25mV ) <Two planar surface > <Two spheres of radious R>
● Particle 이나 surface 사이의 double layer interaction은 exponentially decay 한다. The characteristic decay length = Debye length • ● Small separation에서 counterion이 binding 함으로써 interaction potential에 영향을 끼침 • ●No binding으로 charge density가 일정해지는 영역 • the limiting of small • the limiting pressure • and • ● D가 0에 가까워 지면 potential이나 pressure가 infinite하게 되므로 counterions만이 존재하는 no bulk electrolyte에서도 같은 limiting pressure를 가진다. • General rule for surfaces of constant charge ,all double layer forces tend towards the osmotic limit • ndependent of the bulk electrolyte • ● D가 감소함에 따라 counterion이 binding 해서 P가 limitation밑으로 떨어지면 Posson-Boltzmann 식은 adsorbing ion의 Dissociation constants 를 포함해야 한다.
Charge-regulating interaction의 two main effect • Double layer interaction 의 strength는 constant surface charge 에서의 interaction strength보다 항상 작다. • (Le Chaterlier’s principle) • Two limits 가 존재한다. • Upper(constant surface charge) • Lower(constant surface potential)
12.18 VAN DER WAALS AND DOUBLE-LAYER FORCE ACTING TOGETHER : THE DLVO THEORY ●DLVO theory 로 colloidal의 stability 계산…… Van der waals (Double-layer force와 비교 ): 1. Electrolyte concentration 이나 pH에 둔감 (fixed first approximation) 2. Small distance에서 double layer repulsion 보다 크다 ( power-law interaction) 3. D 0으로 갈때 상대적으로 빨리 증가 ● Ion binding나 salt concentration증가로 double-layer repulsion을 screening함으로써 primary minimum상태의 adhesive contact유도 ●Charge 높고 concentration증가하면 secondary minimum에서 adhere가 일어나지만 repulsive가 쉽게 일어남
12.19 EXPERIMENTAL MEASUREMENTS OF DOUBLE-LAYER AND DLVO FORCES ●Direct measured double-layer and van der Waals forces between two curved mica surface ●DLVO theory is basically sound Continuous curve 는 이론치로 constant charge와 constant potential limits를 보여준다
Significant deviation- non DLVO ● separation below about 8nm the surfaces are closer to each other than the mean distance between the surface charges, and yet the double layer force still behave as if the surface charge were smeared out ● Stern-layer stabilization ; Stern layer로 인해 repulsion이 급격히 증가하여 deep primary minimum을 없애버린다. 그 결과 spontaneous swelling, Reversible coagulation • 그 외에도 ion-correlation, solvation, hydrophobic or steric force 등으로 non-DLVO현상이 일어남
12.20 EFFECTS OF DISCRETE SURFACE CHARGES AND DIPOLES Surface charge density is not uniformly spread out….. Charges on real surfaces are apart ; typically 1-5nm apart on average What effect does this have on the electrostatic interaction between two surface??? Poisson summation formula Charge of opposite sign The field의 x,y 평면에서의 positive or negative charge는 z증가에 따라 빠르게 decay한다. Second lattice of vertical dipoles ; net pressure average는 zero(first approximation)