Trinucleotide repeats (TNRs)
650 likes | 786 Vues
Trinucleotide repeats (TNRs). Dr. Derakhshandeh, PhD. INTRODUCTION. Trinucleotide repeats (TNRs) are microsatellite sequences Disease-causing repeat instability is an important and unique form of mutation linked to more than 40 neurological, neurodegenerative and neuromuscular disorders.

Trinucleotide repeats (TNRs)
E N D
Presentation Transcript
Trinucleotide repeats (TNRs) Dr. Derakhshandeh, PhD
INTRODUCTION • Trinucleotide repeats (TNRs) are microsatellite sequences • Disease-causing repeat instability is an important and unique form of mutation • linked to more than 40 neurological, neurodegenerative and neuromuscular disorders. • I.g. Huntington's disease, myotonic dystrophy and fragile X syndrome
Trinucleotide repeats • TNRs undergo high frequency mutagenesis • To understand better the molecular mechanisms of TNR instability in cultured cells
A new genetic assay was created using a shuttle vector • The shuttle vector contains a promoter-TNR-reporter gene construct whose expression is dependent on TNR length.
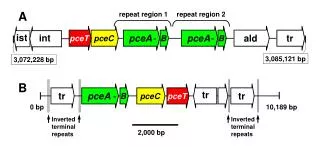
The vector harbors the SV40 ori • (CAG•CTG)25–33
The shuttle vector is propagated in cultured cells • It recovered and analyzed in yeast using selection for reporter gene expression. Richard Pelletier, Nucleic Acids Research 2005 33(17):5667-5676
Disorders caused by trinucleotide repeat • First: the mutant repeats show both somatic and germline instability • Secondly: • an earlier age of onset • and increasing severity of phenotype in subsequent generations (anticipation) • Finally, the parental origin of the disease allele can often influence anticipation • with paternal transmissions carrying a greater risk of expansion for many of these disorders.
Category of the trinucleotide repeat (based on the relative location) • first subclass: • Repeats in non-coding sequences: • For six diseases • second subclass: • Exonic (CAG)n repeats • code for polyglutamine tracts
NON-CODING TRINUCLEOTIDE REPEAT DISORDERS • Large and variable repeat expansions that result in multiple tissue: • dysfunction • degeneration • Phenotypic manifestations within a disease are variable • Degree of somatic heterogeneity
Pre-mutations • The larger mutations often are transmitted from a small pool of clinically silent intermediate size expansions • CGG, GCC, GAA, CTG and CAG • particular trinucleotide sequence + its location with respect to a gene • Important defining factors in dictating the unique mechanism of pathogenesis for each disease
Fragile X syndrome • Fragile X syndrome (FRAXA) • Fragile XE MR (FRAXE) • 1 in 2000 boys1 in 4000 girlsare estimated to be affected
Fragile X Syndrome • most common inherited form of familial mental retardation • (CGG)n trinucleotide expansion in the FMR1 gene leading to the typical Martin-Bell phenotype • Clinical features vary depending on age • Expansion of a (CCG)n repeat in the FMR2 gene corresponds to the FRAXE fragile site • It lies distal to FRAXA • It’s associated with mental retardation, but it is less frequent and lacks a consistent phenotype
The transcription of the FMR1 gene of normal and premutation alleles. Both alleles are translated into FMRP, which is demonstrated by Western blotting (lane N and P). The full mutation allele is hypermethylatedthereforetranscribed, which resultabsence of FMRP (lane F)
Sequence of the 5'-UTR region of the FMR1 gene Sequence of the 5'-UTR region of the FMR1 gene
Fragile X syndrome (FRAXA) • Mental retardation • Macroorchidism • Some dysmorphic features • Hyperactivity
Fragile X syndrome (FRAXA) • expansion of a polymorphic (CGG)n repeat in the 5'-untranslated region (UTR) • > 230 trinucleotides • hypermethylation together with a CpG island within the FMR1 promoter region • transcriptional silencing of the FMR1 gene • reduced FMR1 transcription and loss of gene product (FMRP)
Fragile XE MR (FRAXE) • mild mental retardation • variable behavior abnormalities • expansion of a polymorphic (GCC)n repeat • in the promoter region of the FMR2 gene • the expanded repeats are hypermethylated • leading to transcriptional silencing of FMR2 • subsequent loss of gene product (FMR2)
Friedreich ataxia (FRDA) • autosomal recessive • the only triplet repeat disorder that does not show anticipation • Ataxia (loss of voluntary muscular coordination) • Diminished reflexes • Cardiomyopathy (heart enlargement) • Diabetes • Degeneration in the spinal cord
Friedreich ataxia • FRDA is caused by a large intronic GAA repeat expansion • located on chromosome 9 (Gene:X25/Potein: frataxin) • which leads to reduced gene expression • The expanded AT-rich sequence most probably causes • self-association of the GAA/TTC tract, which stabilizes the DNA in a triplex structure
FRDA & triplex structure • A novel DNA structure • sticky DNA • lengths of (GAA.TTC)n • in intron 1 of the frataxin gene of Friedreich's ataxia patients • Sticky DNA is formed by the association of two purine.purine.pyrimidine (R.R.Y) triplexes • in negatively supercoiled plasmids at neutral pH
Models of structures that may mediate mRNA synthesis and DNA replication inhibition by GAA·TTC repeats
in FRDA patients • (GAA.TTC) (> 59 repeats) • the lengths of (GAA.TTC) (> 59 repeats) • inhibit transcription in vivo and in vitro • adopt the sticky conformation • (GAAGGA.TCCTTC)65 • found in intron 1 • does not form sticky DNA • does not inhibit transcription • or associate with the disease Sakamoto,et al. MMol Cell. 1999 Apr;3(4):465-75.
frataxin is found in the mitochondria of humans • we do not yet know its function • there is a very similar protein in yeast, YFH1, • YFH1 is involved in controlling: • iron levels • and respiratory function • Frataxin and YFH1 are so similar, studying YFH1 may help us understand the role of frataxin in FRDA
Reduced X25 mRNA • decreases frataxin levels • a partial loss of frataxin function • Disruption of the yeast X25 homolog (YFH1): • abnormal accumulation of mitochondrial iron • loss of mtDNA • multiple iron–sulfur-dependent enzyme deficiencies • increased sensitivity to oxidative stress • Frataxin : • hypersensitivity to iron and H2O2 stress
Frataxin insufficiency • frataxin insufficiency may result in abnormal iron–sulfur homeostasis • mitochondrial dysfunction • free radical production • oxidative stress • cellular degeneration Wong, A, et al. Hum. Mol. Genet., 8, 425–430 (1999)
Myotonic dystrophy (DM) • multisystem disorder • highly variable phenotypes • Anticipation • Myotonia • muscle weakness • Developmental abnormalities • mental handicap • Hypotonia • respiratory distress are often evident in the more severe congenital myotonic dystrophy (CDM).
DM • CTG trinucleotide repeat • in the 3'-UTR of the protein kinase gene, DMPK • The CTG repeat is located within the promoter of a upstream homeobox gene • Loss of function of either or both of these proteins could contribute to some of the features in DM Korade-Mirnics, Z. et al. (1998) Nucleic Acids Res., 26, 1363–1368
Spinocerebellar ataxia type 8 (SCBA8) • progressive ataxia • with cerebellar atrophy • decreased brisk reflexes • SCA8 is expressed primarily in the brain • is caused by an expanded CTG repeat in its 3'-terminal exon (~110–250 repeats)
Parkinsonism (PD)
SCA-2 and SCA-3 repeats in Parkinsonism expansion of triplet repeats encoding polyglutamine (polyQ) tracts
POLYGLUTAMINE DISEASES • have repeat expansions that are much smaller in size and variation • characterized by progressive neuronal dysfunction • begins in mid-life and results in severe neurodegeneration
POLYGLUTAMINE DISEASES different polyglutamine diseases have little in common: • the length of the expansion > 35–40 • the greater the number of glutamine repeats in a protein • the earlier the onset of disease and the more severe the symptoms