Homology Modeling via Protein Threading
510 likes | 972 Vues
Homology Modeling via Protein Threading. Kristen Huber ECE 697S Topics in Computational Biology April 19, 2006. Fundamentals of Protein Threading. Protein Modeling Homology Modeling Protein Threading Generalized Overview of a Threading Score

Homology Modeling via Protein Threading
E N D
Presentation Transcript
Homology Modeling via Protein Threading Kristen Huber ECE 697S Topics in Computational Biology April 19, 2006
Fundamentals of Protein Threading • Protein Modeling • Homology Modeling • Protein Threading • Generalized Overview of a Threading Score • Score Methodology based on Multiple Protein Structure Alignment
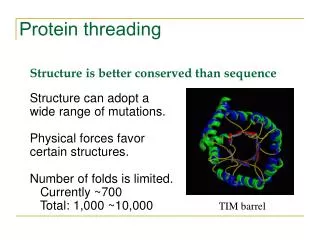
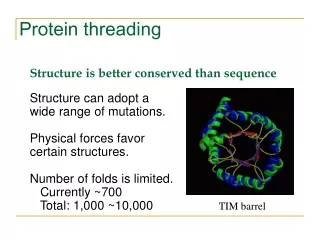
Protein Modeling • 20,000 entries of proteins in the PDB • 1000 - 2000 distinct protein folds in nature • Thought to be only several thousand unique folds in all • Protein Structure Prediction • aim of determining the three-dimensional structure of proteins from their amino acid sequences
Types of Structure Prediction • De novo protein • methods seek to build three-dimensional protein models "from scratch" • Example: Rosetta • Comparative protein • modeling uses previously solved structures as starting points, or templates. • Example: protein threading
Factors that Make Protein Structure Prediction a Difficult Task • The number of possible structures that proteins may possess is extremely large, as highlighted by the Levinthal paradox • The physical basis of protein structural stability is not fully understood. • The primary sequence may not fully specify the tertiary structure. • chaperones • Direct simulation of protein folding is not generally tractable for both practical and theoretical reasons.
Homology Modeling • Homolog a protein related to it by divergent evolution from a common ancestor • 40 % amino-acid identity with its homolog • NO large insertions or deletions • Produces a predicted structure equivalent to that of a medium resolution experimentally solved structure • 25 % of known protein sequences fall in a safe area implying they can be modeled reliably
Homology Modeling Defined • Homology modeling • Based on the reasonable assumption that two homologous proteins will share very similar structures. • Given the amino acid sequence of an unknown structure and the solved structure of a homologous protein, each amino acid in the solved structure is mutated computationally, into the corresponding amino acid from the unknown structure.
Homology Modeling Limitations • Cannot study conformational changes • Cannot find new catalytic/binding sites • Brainstorm lack of activity vs activity • Chymotrypsionogen, trypsinogen and plasminogen • 40% homologous • 2 active, 1 no activity, cannot explain why • Large Bias towards structure of template • Models cannot be docked together
Why Homology Modeling? • Value in structure based drug design • Find common catalytic sites/molecular recognition sites • Use as a guide to planning and interpreting experiments • 70-80 % chance a protein has a similar fold to the target protein due to X-ray crystallography or NMR spectroscopy • Sometimes it’s the only option or best guess
Protein Threading • A target sequence is threaded through the backbone structure of a collection of template proteins (fold library) • Quantitative measure of how well the sequence fits the fold • Based on assumptions • 3-D structures of proteins have characteristics that are semi-quantitatively predictable • reflect the physical-chemical properties of amino acids • Limited types of interactions allowed within folding
Fold Recognition Methods • Derive a 1-D profile for each structure in the fold library and align the target sequence to these profiles • Identify amino acids based on core or external positions • Part of secondary structure • Consider the full 3-D structure of the protein template • Modeled as a set of inter-atomic distances • NP-Hard (if include interactions of multiple residues)
Protein Threading • The word threading implies that one drags the sequence (ACDEFG...) step by step through each location on each template
Position-Specific Iterative BLAST (PSI-BLAST) This program is used to find distant relatives of a protein. First, a list of all closely related proteins is created. These proteins are combined into a general "profile" sequence, which summarises significant features present in these sequences. A query against the protein database is then run using this profile, and a larger group of proteins is found. This larger group is used to construct another profile, and the process is repeated. By including related proteins in the search, PSI-BLAST is much more sensitive in picking up distant evolutionary relationships than a standard protein-protein BLAST.
Generalized Threading Score • Want to correctly recognize arrangements of residues • Building a score function • potentials of mean force • from an optimization calculation. • G(rAB) = kTln (ρAB/ ρAB°) • G, free energy • k and T Boltzmanns constant and temperature respectively • ρ is the observed frequency of AB pairs at distance r. • ρ° the frequency of AB pairs at distance r you would expect to see by chance. • Z-score = (ENat - <Ealt>)/σ Ealt • Natural energies and mean energies of all the wrong structures/ standard deviation
Scoring Different Folds • Goodness of fit score • Based on empirical energy function • Modify to take into account pairwise interactions and solvation terms • High score means good fit • Low score means nothing learned
Some Threading Programs • 3D-pssm (ICNET). Based on sequence profiles, solvatation potentials and secondary structure. • TOPITS (PredictProtein server) (EMBL). Based on coincidence of secondary structure and accesibility. • UCLA-DOE Structure Prediction Server (UCLA). Executes various threading programs and report a consensus. • 123D+ Combines substitution matrix, secondary structure prediction, and contact capacity potentials. • SAM/HMM (UCSC). Basen on Markov models of alignments of crystalized proteins. • FAS (Burnham Institute). Based on profile-profile matching algorithms of the query sequence with sequences from clustered PDB database. • PSIPRED-GenThreader (Brunel) • THREADER2 (Warwick). Based on solvatation potentials and contacts obtained from crystalized proteins. • ProFIT CAME (Salzburg)
Process of 3D Structure Prediction by Threading • Has this protein sequence similarity to other with a known structure? • Structure related information in the databases • Results from threadingprograms • Predicted folding comparison • Threadingon the structure and mapping of the known data • A comparison between the threading predicted structure and the actual one
Protein Threading Based on Multiple Protein Structure AlignmentTatsuya Akutsu and Kim Lan SimHuman Genome Center, Institute of Medical Science, University of Tokyo • NP-Hard if include interactions between 2 or more AA • Determine multiple structural alignments based on pair wise structure alignments • Center Star Method
Center Star Method • Let I0 be the maximum number of gap symbols placed before the first residue of S0 in any of the alignments A(S0; S1); : : : ;A(S0; SN). Let IS0jbe the maximum number of gaps placed after the last character of S0 in any of the alignments, and let Ii be the maximum number of gaps placed between character S0;iand S0;i+1, where Sj:idenotes the i-th letter of string Si • Create a string S0 by inserting I0 gaps before S0, IjSo gaps after S0, andIjgaps between S0;Iand S0;i+1. • For each Sj(j > 0), create a pairwise alignment A(S0; Sj) between S0 and Sj by inserting gaps into Sjso that deletion of the columns consisting of gaps from A(S0; Sj) results in the same alignment as A(S0; Sj). • Simply arrange A(S0; Sj)'s into a single matrix A (note that all A(S0; Sj )'s have the same length).
Simple Threading Algorithm • Apply simple score function based on structure alignment algorithm • Let X = x1……xN (input amino acid sequence) • Ci ( i-th column in A) • Test and analyze results and/or apply constraints
Protein Threading with Constraints • Assume part of the input sequence xi…xi+k must correspond to part of the structure alignment cj…cj+k • Apply constraints
Prediction Power • Entered in CASP3 competition • 17 predictions made • 3 targets evaluated as similar to correct folds • Only team to create a nearly correct model for structure T0043 • Best in competition • 8 evaluated as similar to correct
Next time…. • In depth detail of • Multiple structural alignment program • Multiprospector • Global Optimum Protein Threading with Gapped Alignment • Quality measures for protein threading models • Improvements on threading-based models
Review • Homology Modeling • Based on the reasonable assumption that two homologous proteins will share very similar structures. • Threading • Modeled as a set of inter-atomic distances • NP-Hard (if include interactions of multiple residues) • Build a score function based on energies in order to correctly recognize arrangements of residues • Threading via multiple structural alignment • Score function based upon alignment matrix
Specifics of Protein Threading • Different Threading Types • Multiprospector: Predictions of Protein-Protein Interaction by Multimeric Threading • Global Optimum Protein Threading with Gapped Alignment • Quality measures for protein threading models • Improvements on threading-based models
MULTIPROSPECTOR • An algorithm for the prediction of protein-protein interactions by multimeric threading • Protein–protein interactions are fundamental to cellular function and are associated with processes such as enzymatic activity, immunological recognition, DNA repair and replication, and cell signaling. • Function can be inferred from the nature of the protein with its interactants • Use properties related to the topology of the interface, solvent-accessible surface area and hydrophobicity • Addressed limitations of existing approaches
Method Basis • Thread the sequences through a representative structure template library that, in addition to monomers, also includes each of the chains in representative protein dimer structures. • Compute the interaction energy between a pair of protein chains for those protein structures involved in dimeric complexes. • Stable complex formation determined by the magnitude of the interfacial potentials and the Z-scores of the complex structures relative to that of the monomers.
Interfacial Statistical Potentials • Interfacial pair potentials • P(i, j), (i=1, …, 20; j =1, …,20), • Calculated by examining each interface of the selected dimers • Nobs(i, j) is the observed number of interacting pairs of i, j between two chains. • Nexp(i, j) is the expected number of interacting pairs of i, j {Nexp (i, j) =Xi *Xj * Ntotal} • Apply Boltzman Principal to the ratio to obtain potential of mean force between 2 residues
Multimeric Threading Strategy and Z-Score • Z-score of the score for each probe-template alignment is used to decide if a correct fold is found: • is the standard deviation of energies; Eiis the energy of the i-th sequence of M alternative folds (i 1, …, M).
Global Optimum Protein Threading with Gapped Alignment and Empirical Pair Score Functions • The structural model corresponds to an annotated backbone trace of the secondary structure segments in the conserved core fold. • Loops are not considered part of the conserved fold, and are modeled by an arbitrary sequence-specific loop score function. • Alignment gaps are confined to the connecting non-core loop regions • Each distinct threading is assigned a score by an assumed score function • Exponentially large search space of possible threadings • NP-hard search spaces as large as 9.6x1031 at rates ranging as high as 6.8 x1028 equivalent threadings per second
Gapped Protein Threading Methodology • Common core of four secondary structure segments • Spatial interactions. Small circles represent amino acid residue positions (core elements), and thin lines connect neighbors in the folded core. • Thread through model by placing successive sequence amino acid residues into adjacent core elements. Tax indexes the sequence residue placed into the first element of segment X. Sequence regions between core segments become connecting turns or loops. • Sets used in the branch-and-bound search are defined by lower and upper limits (dark arrows, labeled baxand daxfor segment X)
General Pairwise Score Function • For any threading t, let fv(v, t) be the score assigned to core element or vertex v • fe({u, v}, t) the score assigned to interaction or edge {u, v} • f1(λi , t) the score assigned to loop region λi • Then the total score of the threading is: • Rewrite function of threading pairs of core segments
Branch-and-Bound Search Algorithm • branch-and-bound search requires the ability to • represent the entire search space as a set of possibilities • split any set into subsets • compute a lower bound on the best score achievable within any subset • After some finite number of steps, the chosen set will contain only one threading (equals its lower bound)
Splitting the Search Space • The set of all legal threadings is represented by the hyper-rectangle • lower bound on the score f(t) attainable by any threading t in the set T • summing lower bounds on each term separately The enclosing mintЄTensures that the lower bound will be instantiated on a specific legal threading tlbЄT. This will be used in splitting T, below. The equation further ensures that the singleton term, in g1(i, ti), remains consistent both with the terms that reflect loop scores, in g2(i - 1, i, ti-1, ti), and with the other (non-loop) pairwise terms, in g2(i, j, ti, uj). The inner minuЄTallows a different vector u for each i, but requires u to be a legal threading.
Quality Measures for Protein Threading Models • Evaluation of different prediction methods for protein threading • Purpose: • determine if one method to build a model is better than another • optimize the performance of existing methods. • Threading Assessment: • ability to predict the correct fold • the similarity of the model to the correct structure
Methods of Comparison Defined • Global • consider all residues in both the model and the correct structure in an "alignment dependent“ fashion • Alignment Dependent • based on an exact match between the residues in the model and the correct structure • Alignment Independent • based on a structural superposition between the model and the correct structure • Template Based • available for models that are created from the sequence being aligned onto a single structural template.
Comparison Results • Most methods correlate to each other • 0.51 model-normalized • 0.41 template-normalized • High quality homology-models correlate less with the rest of the data • Measures of same type correlate well and tend to cluster
A Need for Improvement • Resulting models obtained from threading approaches are usually of very low quality, with gaps and insertions in threading alignments that somehow have to be connected or closed • Various threading methods and their associated scoring functions only focus on aspects of protein structure and a subset of their possible interactions.
Method of Improvement • Employs a lattice model • SICHO (Side Chain Only) • The model has been refined by incorporating evolutionary information into the interaction scheme. • a Monte Carlo annealing procedure attempts to find a conformation that maintains some (but not all) features of the original template • optimizes packing and intra-protein interactions
Lattice Model • The model chain consists of a string of virtual bonds connecting the interaction centers that correspond to the center of mass of the side chains and the backbone alpha carbons. • These interaction centers are projected onto an underlying cubic lattice with a lattice spacing of 1.45 A° • A cluster of excluded volume points is associated with each bead of the model chain. • Each cluster consists of 19 lattice points • Closest approach distance from another cluster labels smallest inter-residue distance
Interaction Scheme • Starting Model takes on a tube form • Energy potentials. • generic, sequence-independent, biases that penalize against non protein-like conformations • two-body and multibody potentials extracted from a statistical analysis of known protein structures. • Evolutionary information extracted from multiple sequence alignments. • The stiffness/secondary structure bias term has the following form: • Estiff= -Єgen [Σ min{0.5, max (0, wi●wi+2)}] - Єgen [Σ min{0.5, max (0, wi●wi+4)}]
Interaction Scheme • A weak bias being introduced towards helix-type and beta-type expanded states • Estruct= Σ{δH1(i) + δ H2(i) + δ E1(i) + δ E2(i)} • δ H1 and δ H2 contributions defined as a broad range of helical/turn conformations • δ E1 and δ E2 as expanded conformations • Generic packing interactions • Short range interactions • Pairwise Interactions • Multi-body Interactions • statistical potential for residue type A having np parallel and na anti-parallel contacts. • Emulti= ΣEm(A,np,na) • Total energy • Etotal = Estiff + Emap + 0.875EH-bond + 0.75Eshort + 1.25Epair + 0.5Esurface + 0.5Emulti
Threading Model Refinement • a) Generate the threading alignment between the unknown sequence and the template structure. • b) Derive the sequence similarity-based short and long range pairwise potentials. • multiple alignments with homologous sequences of unknown structures were used in the potential derivation procedures.) • c) Build the starting continuous model chain onto the lattice-projected template structure. • d) Build the tube around the aligned fragments of the template structure. Then, perform the first stage of Monte Carlo refinement. • e) Refinement of the structure • assume to be the new template • Narrow restraints • Select lowest energy structures • All atom models using MODELLER.24