Download

1 / 25
250 likes | 461 Vues
RETROSPECTIVE EVALUATION OF THE PATIENTS WITH CYSTIC FIBROSIS. DR.LALE PULAT SEREN ZEYNEP KAMİL MATERNITY AND CHILDREN’S TRAINING AND RESEARCH HOSPITAL. CYSTIC FIBROSIS. The most common life-limiting inherited disorder Autosomal recessive genetic trait

E N D
RETROSPECTIVE EVALUATION OF THE PATIENTS WITH CYSTIC FIBROSIS DR.LALE PULAT SEREN ZEYNEP KAMİL MATERNITY AND CHILDREN’S TRAINING AND RESEARCH HOSPITAL
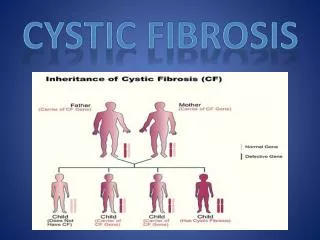
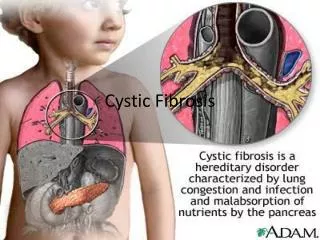
CYSTIC FIBROSIS • The most common life-limiting inherited disorder • Autosomal recessive genetic trait • Mutation of CF transmembrane regulator (CFTR) gene • The most prevalent one among 1340 different mutations of CFTR is ΔF508 • Variable involvement of the lungs, pancreas, intestinal tract, liver and sweat glands are presented.
Chronic sinopulmonary disease Chronic cough and sputum Wheezing Nasal polyp Digital clubbing • Gastrointestinal abnormalities Intestinal-Diarrhea,obstruction Liver - Chronic liver disease, cirrhosis Nutrition – growth retardation ●Male genitourinary abnormalities Infertility
CF • Incidence 1 / 2500 – 3500 in whites 1 / 2000 – 4000 in Turkish population (Gürson et al., 1973) 1000 –1500 patients are estimated to be followed up for CF in Turkey.
Material and Methods • 23 CF patients followed between 1998-2003 were evaluated retrospectively • Diagnosis was confirmed at least two sweat test positivity (Cl > 60mEq/L) • Pilocarpine iontophoresis was used for sweat testing.
Material and Methods • 10 female ( 43,4 %), 13 male (56,6 %) ( 23 patients ) ● 2 patients were died (1 from hypovolemic shock due to gastroenteritis and 1 from cardiopulmonary insufficiency due to lung disease)
Results • Age range of the patients 45 days – 11 months ( median 5,5 months ) • Median age at the time of diagnosis 2,5 months -12 months ( median 6,4 months )
Results • Consangineous marriage 66,2 % ( n = 15 ) • History of death sibling 21,7 % ( n = 5 )
Results • The most common causes of hospitalization; - Recurrent pulmonary infection - Gastroenteritis - Growth retardation
Nasopharyngeal aspirate cultures were available in 10 patients and the identified bacterias were; S. aureus 2 cases P. aeruginosa 2 cases H. influenza 1 case □ Klebsiella sp. were grown in hemocultures of 2 patients.
The diagnosis of CF is generally made in the first year of life. • Abnormalities in the electrolyte content of sweat is presented from birth to death. • More than 60 mEq/L of chloride in sweat is pathologic and diagnostic for CF but threshold levels of 40 mEq/L for infants (esp. younger than 3 months) have been suggested. Beauchamp M. et al. Sweat Testing. Pediatr Pulmonol 2005;39:507-11
Mutational heterogeneity and environmental factors appear responsible for highly variable organ involvement. • Involvement of the respiratory tract is responsible 90 % of morbidity and mortality.
The most common sign of GIS is exocrine pancreatic insufficiency related steatorrhea. • Malabsorbtion of fats and proteins are predominated.
CF patients required a multidisciplinary treatment protocol. • Chest physical therapy is the most important part of the treatment. • Inhaled hypertonic saline, N-acetylcysteine and DNase are used for the removal of viscouse secretions. • Culture appropiate antibiotics have to be choosen for acute exacerbations of chronic lung disease.
Conclusion • Life quality in CF has been progressively increased with early diagnosis and modern treatment methods, in recent years. • Since, consangineous marriages are common in our country, to form a standart national work-up programme is required.