Heterogeneous Catalysis 6 lectures
Heterogeneous Catalysis 6 lectures. Dr. Adam Lee Surface Chemistry & Catalysis Group. Synopsis.


Heterogeneous Catalysis 6 lectures
E N D
Presentation Transcript
Heterogeneous Catalysis 6 lectures Dr. Adam Lee Surface Chemistry & Catalysis Group
Synopsis Heterogeneous Catalysis is crucial to diverse industries ranging from fuels to food and pharmaceuticals. This course will introduce a wide range of heterogeneous catalysts and associated industrial processes. Methods for the preparation, characterisation and testing of solid catalysts will be discussed. Fundamentals of surface reactions and catalyst promotion are addressed, and finally some applied aspects of catalyst reactor engineering will be considered. Topics: • Heterogeneous catalysts: definitions, types, advantages • Catalyst surfaces: adsorption processes, kinetics • Structure-sensitivity: dispersion, active site • Bimetallic catalysts: selectivity control • Catalyst preparation • Catalyst characterisation Recommended Texts: • Basis and Applications of Heterogeneous Catalysis: Mike Bowker,Oxford Primer, (1998) • Catalytic Chemistry: B.C.Gates, Wiley (1992) • Heterogeneous Catalysis: G.C.Bond OUP 2nd Ed (1987)
Lecture 1 Overview • What are catalysts and why are they beneficial ‘Why haven’t they been used more widely when so many examples in petrochemical industry?’ • Types of catalysts • Properties of catalysts • Calculation of TON & measurement of kinetic parameters • Overview of typical classes of reactions and catalysts used • Environmental considerations
How can we accelerate a chemical reaction? Organic Chemistry (1805) Physical Chemistry Discovery of Catalysis (1835) - Petrochemical & oil refining industry recognise promise - Catalytic technology generates >10 trillion $/yr - Clean technology (1990?) - applications in plastics, fabrics, food, fuel Why don’t we use a catalyst? Use reagents - stoichiometric - separation problems - TOXIC waste - Industrial fine chemicals processes developed - Carry on using reagents
Permanganate, Manganese dioxide, Chromium (VI) (<0.10 ppm) Metal Hydrides, (NaBH , LiAlH ) 4 4 Reducing metals ( Na, Fe, Mg, Zn) Potassium butoxide, diisopropylamine Tetramethyl guanidine AlCl , BF , ZnCl , H SO 3 3 2 2 4 Typical Reagents Oxidation Reduction Basic reagents Acidic reagents C-C Coupling Homogeneous Pd based complexes
Nobel Prize in Chemistry 2007 – Gerhard Ertl Importance of Heterogeneous Catalysis Chemicals Industry: >90% of global chemical output relies upon heterogeneous catalysed processes Economics: • ~20% of world GNP dependent on processes or derived products • Equates to $10,000 billion/year!! Environment: • Ozone depletion catalysed over aerosol surfaces in Polar Stratospheric Clouds • Pollution control (catalytic converters, VOC destruction) • Clean synthesis (waste minimisation, benign solvents, low temperature) • Power generation
HDPE LDPE Polymerisation (1957/1991) nC2H2 Zeigler-Natta /Metallocene Catalytic Cracking (1964) CxH2x+2 Cx-2H2x-2 CxH2x+2 Cx-2H2x-4 Faujasitic zeolites Historical Evolution
HC + CO + NOX CO2 + H2O + N2 Pt/Rh/Al2O3 Chiral pocket Automotive Emission Control (1976) Chiral Catalysis (1988)
Advantages of Catalytic Technology ‘A catalyst is a material that enhances the rate and selectivity of a chemical reaction without itself being consumed in the reaction.’ Swedish Chemist - Jöns Jakob Berzelius (1779-1848) Minimize FEEDSTOCK and reduce ENERGY costs More efficient use of raw materials.
Classes of Catalyst • Heterogeneous - active site immobilised on solid support - tuneable selectivity - easily separated • Homogeneous - organometallic complexes widely used - more active than heterogeneous, - high selectivity - difficult to separate • Bio-catalysts - enzymes, bacteria, fungi - highly selective • Phase transfer - Reagent soluble in separate phase to substrate - use PTC to transfer reagent into organic
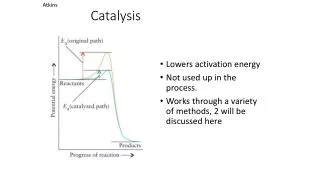
kforward Reactants Products kback Catalyst Definitions Catalyst: a material that enhances the rate and selectivity of a chemical reaction without itself being consumed in the reaction. Rates (kinetics): Rate = rate constant x [reactant]n Rate constant (k or k’) = A exp (-EAct/RT) Consider, All catalysts work by providing alternative pathways: • different, lower EAct • accelerates both forward AND reverse reactions (increase kf and kb) • catalysts do not influence how MUCH product forms
Uncatalysed Catalysed Catalyst Definitions Energetics: Reactants do not all have same energy: Boltzmann distribution So what determines theoretical product yield?? - thermodynamic driving force, G = -nRT ln(K) Large –ve G large +ve ln(K) huge K ~100 % Yield http://www.chemguide.co.uk/physical/basicrates/catalyst.html#top Catalysts do not affect K!
Catalyst Definitions Goal of catalytic research is improved activity & selectivity Alter rate constants: k For simple reax. A B + C • Activity = • Selectivity= = Yield of Desired Productx 100% Total Yield of all Product mol . s-1 rate of reaction %relative formation of specific product
Triglyceride transesterification Catalyst Efficiency: 1 Conversion • The % of reactant that has reacted Conversion = (Amt of Reactant at t0) - (Amt of Reactant at t1) x 100 (Amt of Reactant at t0) Activity = -d[Tributyrin] = 20 = 1 mmol.s-1 dt 20 Conversion = 20 % Biodiesel
Triglyceride transesterification [FAME] [Diglyceride]+[Monoglyceride]+[FAME] 45 20+10+45 x 100 = x 100 Tri-glyceride Methyl-butanoate (FAME) Di-glyceride Selectivity to FAME? = 60 %
Catalyst Efficiency: 2 Reagents are often stoichiometric - single use • By definition catalysts must be regenerated once product formed. • Need a parameter to compare efficiency of catalysts. Turn over number (TON)- Number of reactions a single site can achieve e.g. 1 mmol Pd converts 1000 mmols of COCO2 Turn over frequency (TOF)- Number of reactions per site per unit time. e.g. 1 mmol Pd converts 1000 mmols of COCO2 in 10 s To be valid TOF must be measured in absence of: - mass transport limitations - deactivation effects TON = 1000 TOF = 100 s-1
Catalyst Constituents Active Phase - transition-metal - highly dispersed - reduced/oxidic/sulphided state ‘Inert’ Support - high surface area oxide - high porosity - high thermal/mechanical stability Sn - Naptha reforming Cl - Ethylene epoxidation K2O - NH3 synthesis C - Catalytic cracking S, Pb - Car exhaust catalysts
Active Component Responsible for the principal chemical reaction Features: • activity, selectivity, purity • surface area, distribution on support, particle size Types: • Metals • Semiconductor oxides and sulphides • Insulator oxides and sulphides Platinum particles on a porous carbon support Transmission Electron Micrograph
Support Main function is to maintain high surface area for active phase Other features include: • porosity • mechanical properties • stability • dual functional activity • modification of active component Types: • high melting point oxides (silica, alumina) • clays • carbons
Advantages and Limitations of Heterogeneous Catalysts • Ease of removal from reaction and possible to recycle • Diffusional effects - reaction rates may be limited by diffusion into/out of pores. • May need to re-optimise plants (often batch reactors) for solid-liquid processes - separation technology • Opportunity to operate continuous processes
Why the Implementation Delay?? Apathy - Fine chemicals synthesis often on small scale, magnitude of waste not appreciated. Cost - Conventional reagents are cheap, catalysts require development………(i.e. Investment!) Time - Fine chemicals have a short life cycle compared to bulk chemicals:‘Time to market’ is critical. ‘…classical methods are broadly applicable and can be implemented relatively quickly. ..…the development of catalytic technology is time consuming and expensive.’ R.A.Sheldon & H.Van Bekkum - Eds. Fine chemicals through heterogeneous catalysis
Dr. Paul Anastas Director of Green Chemical Inst. Washington D.C. ex. White House Asst. Director for Environment
“It is better to prevent waste than to treat or clean up waste after it is formed” Chemical Process No waste
Only required product C + D + E + F ... A + B C (only product) “Synthetic methods should be designed to maximise the incorporation of all materials used into the final product” Selectivity
Filtration “Energy requirements should be recognised for their environmental impacts and minimised. Synthetic methods should be conducted at ambient pressure and temperature” High Activity Heating Cooling Stirring Distillation Compression Pumping Separation Energy requirement (electricity) Global warming Burn fossil fuel CO2 to atmosphere
“Unnecessary derivatisation (blocking group, protection/deprotection..) should be avoided wherever possible” Selectivity
CONCLUSION: “Selective catalysts are superior to stoichiometric reagents” Stoichiometric 4-Chlorobenzophenone Catalytic
Catalysis in Action: C2H2 on Pd(111) Scanning Tunnelling Microscope movie - real-time molecular rotation Further Info Even More Info!
Lecture 3/4 Overview • Reaction kinetics and diffusion limitations • Langmuir adsorption isotherm • Unimolecular reaction • Bimolecular reactions • Surfaces
Kinetics of Catalysed Reactions • Kinetics of heterogeneously catalysed liquid phase reactions are largely governed by diffusion limitation within the porous solid • Require a new range of heterogeneous catalysts tailored for liquid phase organic reactions offering... - pore structure - ease of separation - high activity - high selectivity to desired products.
Comparison Batch Reactor Batch/Flow Reactor
Key Considerations • Diffusional effects - (Mass Transfer) • Adsorption strength - • Mechanism - • Heat transfer - Solvent polarity Ratio of reactant Competitive adsorption Adsorption of product/by products (e.g. H2O) Site blocking Solvent adsorption Study rate as function of concentration and compare theoretical profile Hot spots? In exothermic reactions rapid removal of heat from active site is essential
Reactant film O2 Diffusion Parameters k1 k7 k2 k6 k3 k4 k5 Porous catalyst structure A B k1 = Film mass transfer to ext. surface k2 = Diffusion into Catalyst Pore (Bulk or Knudsen Diffusion) k3 = Adsorption on surface k4 = Reaction k5 = Desorption of Product k6 = Diffusion of Product k7 = Film mass transfer away ext. surface Reax. Mix Gas diffusion kinetics important in liquid oxidation/hydrogenation - high pressure needed to increase solubility
Henry’s Law Dissolution is EXOTHERMIC For dissolution of oxygen in water, O2(g) <--> O2(aq), enthalpy change under standard conditions is -11.7 kJ/mole. Raise PRESSURE Not temperature
Arrhenius const Activation Energy Diffusion control ln kapp Reaction control 1/T Activation Energy - Diffusion Limitation? • At low T reaction processes dominate • At high T diffusional effects become rate limiting • Typical Arrhenius plot kapp = Aexp (-Eapp/RT) lnkapp = LnA - Eapp/RT
Test for Diffusion Limitation • Rate [Cat]n n=1 if no diffusion limitation • Rate with agitation, or gas flow • Eapp is low 10-15 kJmol-1 Diffusional Step Chemical Step Small T dep (T1/2 or T3/2) High T dep Ea ~ 20-200kJmol-1
Surface Terminology • Substrate (adsorbent) - the solid surface where adsorption occurs Adsorbate - the atomic/molecular species adsorbed on the substrate
Adsorbed NH3 reacting over Fe = 1 Langmuir Adsorption Isotherm • Adsorption - the process in whichspecies ‘bind’ to surface of another phase • Coverage - the extent of adsorption of a species onto a surface ()
GAS/LIQUID reactants, products solvents CATALYST absorbate Langmuir Adsorption Isotherm:refresher • Predicts adsorbate coverage () calculate reaction rates optimise reaction conditions (T, pressure) • Chemical equilibria exist during all reactions - stabilities of adsorbate vs. gas/liquid - temperature (surface and reaction media) - pressure (liquid conc.) above catalyst
S* + M S----M [S----M] adsorbate coverage [S*] vacancies (1- ) Reactants [M] gas pressure P Products Equilibrium between the gas molecules M, empty surface sites S and adsorbates e.g. for non-dissociative adsorption Assumption 1: Fixed number of identical, localised surface sites
b Langmuir Adsorption Isotherm Equilibrium constant, b is Rearrange in terms of , - b called sticking-probability and depends on Hads Assumption 2: Hads and thus b is temperature & pressure independent
Unimolecular Decomposition Consider the surface decomposition of a molecule A , i.e. A (g)« A (ads)® Products Let us assume that : • decomposition occurs uniformly across surface sites (not restricted to a few special sites) • products are weakly bound to surface and, once formed, rapidly desorb • the rate determining step (rds) is the surface decomposition step Under these circumstances, the molecules of A on the surface are in equilibrium with those in the gas phase predict surface conc. of A from Langmuir isotherm Assumption 3: Hads is coverage independent Assumption 4: Only 1 adsorbate per site q = b.P / ( 1 + b.P )
Rate of surface decomposition (reaction) is given by an equation: Rate = k q (assuming that the decomposition of Aads occurs in unimolecular elementary reaction step and that kinetics are 1st order in surface concentration of intermediate Aads) Substituting for the q gives us equation for the rate in terms of gas pressure above surface Two extreme cases: • Limit 1 :b.P << 1 ; i.e. a 1st order reaction (with respect to A) with an 1st order rate constant , k' = k.b . This is low pressure (weak binding) limit : Rate = k b P / ( 1 + b P ) and Rate ~ k.b.P then ( 1 + b.P ) ~ 1 steady state surface q of reactantv. small
Limit 2 : b.P >> 1 ; then ( 1 + b.P ) ~ b.P and Rate ~ ki.e. zero order reaction (with respect to A) This is the high pressure (strong binding) limit : steady state surface q of reactant ~100% Rate shows the same pressure variation as q (not surprising since rate q!) Rate = k b P / ( 1 + b P )
Bimolecular Reactions:1 Langmuir-Hinshelwood type reaction : Assume that surface reaction between two adsorbed species is the rds. If both molecules are mobile on the surface and intermix then reaction rate given by following equation for bimolecular surface combination step: Rate = k qA qB Since q = b.P / ( 1 + b.P ), when A& B are competing for same adsorption sites the relevant equations are: A (g) A (ads) B (g) B (ads) A (ads)+ B (ads)AB (ads)AB (g) rds fast
Competitive Adsorption Rate Pure A Pure B [A]/[B] Substituting these into the rate expression gives : Look at several extreme limits: • Limit 1 : bAPA<< 1 & bBPB<< 1 In this limit qA & qB are both very low , and Rate®k . bAPA . bBPB= k' . PA. PB1st order in both reactants • Limit 2 :bAPA<< 1 <<bBPB In this limit qA® 0 , qB® 1 , and Rate ® k . bA PA/ (bB PB ) = k' . PA/ PB q = b.P / ( 1 + b.P ) 1st order in A negative 1st order in B
Rate = k qA [B] Rate [A ]/ [B] Bimolecular Reactions:2 Eley-Rideal type reaction : Consider same chemistry A (g) A (ads) A (ads)+ B (gas)AB (ads)AB (gas) last step is direct reax between adsorbed A* and gas-phase B. A + B AB rds fast A varied where [B] is pressure/conc in gas or liquid phase
However Without extra evidence cannot conclude above reaction is Eley-Rideal mechanism… last step may be composite and consist of the following stages B (g) B (ads) A (ads)+ B (ads)AB (ads)AB (g) with extremely small steady-state coverage of adsorbed B Test by monitoring rate • vary qA • vary ratio of or over wide range slow fast fast Langmuir-Hinshelwood not Eley-Rideal. need free sites
O CO Calculated energy diagram Example 1 Langmuir-Hinshelwood: CO oxidation over Pt Highest rate of CO2 production under slightly oxidising conditions: - a high concentration (~0.75 monolayer) of surface O - significant no. of Oa vacancies (empty sites) - CO adsorbs in vacancy with only small energy barrier CO(g)+½O2(g) CO(g)+O(a) Reaction pathway