Download

1 / 44
440 likes | 564 Vues
This paper explores the methodologies for protein sequence alignment utilizing Pair Hidden Markov Models (HMMs) and dynamic programming approaches. It delves into affine gap alignment, the evolution of proteins, and the significance of their sequences and structures. A detailed examination of actin as a model protein underscores its role in cellular processes. Additionally, it presents insights into protein classification and phylogenetic relationships, alongside the challenges in predicting protein structure. Key computational methods used in structure determination are also highlighted.

E N D
A state model for alignment M (+1,+1) Alignments correspond 1-to-1 with sequences of states M, I, J I (+1, 0) J (0, +1) -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC--- TAG-CTATCAC--GACCGC-GGTCGATTTGCCCGACC IMMJMMMMMMMJJMMMMMMJMMMMMMMIIMMMMMIII
Let’s score the transitions s(xi, yj) M (+1,+1) Alignments correspond 1-to-1 with sequences of states M, I, J s(xi, yj) s(xi, yj) -d -d I (+1, 0) J (0, +1) -e -e -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC--- TAG-CTATCAC--GACCGC-GGTCGATTTGCCCGACC IMMJMMMMMMMJJMMMMMMJMMMMMMMIIMMMMMIII
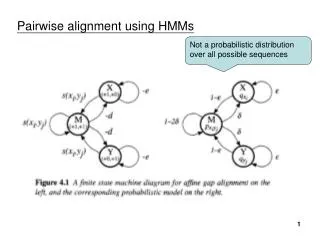
Alignment with affine gaps – state version Dynamic Programming: M(i, j): Optimal alignment of x1…xi to y1…yjending in M I(i, j): Optimal alignment of x1…xi to y1…yj ending in I J(i, j): Optimal alignment of x1…xi to y1…yjending in J The score is additive, therefore we can apply DP recurrence formulas
Alignment with affine gaps – state version Initialization: M(0,0) = 0; M(i, 0) = M(0, j) = -, for i, j > 0 I(i,0) = d + ie; J(0, j) = d + je Iteration: M(i – 1, j – 1) M(i, j) = s(xi, yj) + max I(i – 1, j – 1) J(i – 1, j – 1) e + I(i – 1, j) I(i, j) = max d + M(i – 1, j) e + J(i, j – 1) J(i, j) = max d + M(i, j – 1) Termination: Optimal alignment given by max { M(m, n), I(m, n), J(m, n) }
Brief introduction to the evolution of proteins Protein sequence and structure Protein classification Phylogeny trees Substitution matrices
Actin sequence • Actin is ancient and abundant • Most abundant protein in cells • 1-2 actin genes in bacteria, yeasts, amoebas • Humans: 6 actin genes • -actin in muscles; -actin, -actin in non-muscle cells • ~4 amino acids different between each version MUSCLE ACTIN Amino Acid Sequence 1 EEEQTALVCD NGSGLVKAGF AGDDAPRAVF PSIVRPRHQG VMVGMGQKDS YVGDEAQSKR 61 GILTLKYPIE HGIITNWDDM EKIWHHTFYN ELRVAPEEHP VLLTEAPLNP KANREKMTQI 121 MFETFNVPAM YVAIQAVLSL YASGRTTGIV LDSGDGVSHN VPIYEGYALP HAIMRLDLAG 181 RDLTDYLMKI LTERGYSFVT TAEREIVRDI KEKLCYVALD FEQEMATAAS SSSLEKSYEL 241 PDGQVITIGN ERFRGPETMF QPSFIGMESS GVHETTYNSI MKCDIDIRKD LYANNVLSGG 301 TTMYPGIADR MQKEITALAP STMKIKIIAP PERKYSVWIG GSILASLSTF QQMWITKQEY 361 DESGPSIVHR KCF
Protein Phylogenies • Proteins evolve by both duplication and species divergence
Structure Determines Function The Protein Folding Problem • What determines structure? • Energy • Kinematics • How can we determine structure? • Experimental methods • Computational predictions
Primary Structure: Sequence • The primary structure of a protein is the amino acid sequence
Primary Structure: Sequence • Twenty different amino acids have distinct shapes and properties
Primary Structure: Sequence A useful mnemonic for the hydrophobic amino acids is "FAMILY VW"
Secondary Structure: , , & loops • helices and sheets are stabilized by hydrogen bonds between backbone oxygen and hydrogen atoms
PDB Growth New PDB structures
Protein classification • Number of protein sequences grows exponentially • Number of solved structures grows exponentially • Number of new folds identified very small (and close to constant) • Protein classification can • Generate overview of structure types • Detect similarities (evolutionary relationships) between protein sequences • Help predict 3D structure of new protein sequences Classification of 25,973 protein structures in PDB Morten Nielsen,CBS, BioCentrum, DTU
Protein world Protein structure classification Protein fold Protein superfamily Protein family Morten Nielsen,CBS, BioCentrum, DTU
Structure Classification Databases • SCOP • Manual classification (A. Murzin) • scop.berkeley.edu • CATH • Semi manual classification (C. Orengo) • www.biochem.ucl.ac.uk/bsm/cath • FSSP • Automatic classification (L. Holm) • www.ebi.ac.uk/dali/fssp/fssp.html Morten Nielsen,CBS, BioCentrum, DTU
Major classes in SCOP • Classes • All a proteins • All b proteins • a and b proteins (a/b) • a and b proteins (a+b) • Multi-domain proteins • Membrane and cell surface proteins • Small proteins • Coiled coil proteins Morten Nielsen,CBS, BioCentrum, DTU
All a: Hemoglobin (1bab) Morten Nielsen,CBS, BioCentrum, DTU
All b: Immunoglobulin (8fab) Morten Nielsen,CBS, BioCentrum, DTU
a/b:Triosephosphate isomerase (1hti) Morten Nielsen,CBS, BioCentrum, DTU
a+b: Lysozyme (1jsf) Morten Nielsen,CBS, BioCentrum, DTU
Families • Proteins whose evolutionarily relationship is readily recognizable from the sequence (>~25% sequence identity) • Families are further subdivided into Proteins • Families are divided into Species • The same protein may be found in several species Fold Superfamily Family Proteins Morten Nielsen,CBS, BioCentrum, DTU
Superfamilies • Proteins which are (remotely) evolutionarily related • Sequence similarity low • Share function • Share special structural features • Relationships between members of a superfamily may not be readily recognizable from the sequence alone Fold Superfamily Family Proteins Morten Nielsen,CBS, BioCentrum, DTU
Folds • >~50% secondary structure elements arranged in the same order in sequence and in 3D • No evolutionary relation Fold Superfamily Family Proteins Morten Nielsen,CBS, BioCentrum, DTU
Substitutions of Amino Acids Mutation rates between amino acids have dramatic differences!
Substitution Matrices BLOSUM matrices: • Start from BLOCKS database (curated, gap-free alignments) • Cluster sequences according to > X% identity • Calculate Aab: # of aligned a-b in distinct clusters, correcting by 1/mn, where m, n are the two cluster sizes • Estimate P(a) = (b Aab)/(c≤d Acd); P(a, b) = Aab/(c≤d Acd)
Probabilistic interpretation of an alignment An alignment is a hypothesis that the two sequences are related by evolution Goal: Produce the most likely alignment Assert the likelihood that the sequences are indeed related
A Pair HMM for alignments Model M 1 – 2 This model generates two sequences simultaneously Match/Mismatch state M: P(x, y) reflects substitution frequencies between pairs of amino acids Insertion states I, J: P(x), P(y) reflect frequencies of each amino acid : set so that 1/2 is avg. length before next match :set so that 1/(1 – ) is avg. length of a gap M P(xi, yj) 1 – 1 – I P(xi) J P(yj) optional
A Pair HMM for unaligned sequences Model R Two sequences are independently generated from one another P(x, y | R) = P(x1)…P(xm) P(y1)…P(yn) = i P(xi) j P(yj) 1 1 J P(yj) I P(xi)
To compare ALIGNMENT vs. RANDOM hypothesis 1 – 2 Every pair of letters contributes: M • (1 – 2) P(xi, yj) when matched • P(xi) P(yj) when gapped R • P(xi) P(yj) in random model Focus on comparison of P(xi, yj) vs. P(xi) P(yj) M P(xi, yj) 1 – 1 – I P(xi) J P(yj) 1 1 J P(yj) I P(xi)
To compare ALIGNMENT vs. RANDOM hypothesis Idea: We will divide alignment score by the random score, and take logarithms Let P(xi, yj) s(xi, yj) = log ––––––––– + log (1 – 2) P(xi) P(yj) (1 – ) P(xi) d = – log ––––––––––––– (1 – 2) P(xi) P(xi) e = – log –––––– P(xi) =Defn substitution score =Defn gap initiation penalty =Defn gap extension penalty
The meaning of alignment scores • The Viterbi algorithm for Pair HMMs corresponds exactly to global alignment DP with affine gaps VM(i, j) = max { VM(i – 1, j – 1), VI( i – 1, j – 1) – d, Vj( i – 1, j – 1) } + s(xi, yj) VI(i, j) = max { VM(i – 1, j) – d, VI( i – 1, j) – e } VJ(i, j) = max { VM(i – 1, j) – d, VI( i – 1, j) – e } • s(.,.) ~how often a pair of letters substitute one another • 1/mean length of next gap • 1/mean arrival time of next gap
The meaning of alignment scores Match/mismatch scores: P(xi, yj) s(a, b) log –––––––––– (ignore log(1 – 2) for the moment) P(xi) P(yj) Example: DNA regions between human and mouse genes have average conservation of 80% • What is the substitution score for a match? P(a, a) + P(c, c) + P(g, g) + P(t, t) = 0.8 P(x, x) = 0.2 P(a) = P(c) = P(g) = P(t) = 0.25 s(x, x) = log [ 0.2 / 0.252 ] = 1.68 • What is the substitution score for a mismatch? P(a, c) +…+P(t, g) = 0.2 P(x, yx) = 0.2/12 = 0.167 s(x, y x) = log[ 0.167 / 0.252 ] = -1.42 • What ratio matches/(matches + mism.) gives score 0? 1.67/(1.67+1.42) = 54 % (~halfway between random and conserved model)
Substitution Matrices BLOSUM matrices: • Start from BLOCKS database (curated, gap-free alignments) • Cluster sequences according to > X% identity • Calculate Aab: # of aligned a-b in distinct clusters, correcting by 1/mn, where m, n are the two cluster sizes • Estimate P(a) = (b Aab)/(c≤d Acd); P(a, b) = Aab/(c≤d Acd)
BLOSUM matrices BLOSUM 50 BLOSUM 62 (The two are scaled differently)