Download

1 / 27
380 likes | 813 Vues
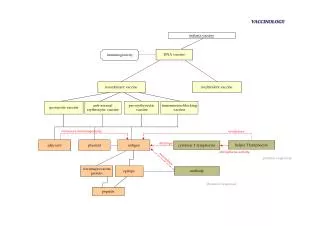
Computational Vaccinology. Darren R Flower. http://www.jenner.ac.uk/res-bio.htm darren.flower@jenner.ac.uk. WHOLE ORGANISM. SUBUNIT VACCINE. EPITOPE VACCINE. C OMPUTATIONAL V ACCINOLOGY. Vaccines induce protective immunity, an enhanced adaptive immune response to re-infection.

E N D
Computational Vaccinology Darren R Flower http://www.jenner.ac.uk/res-bio.htm darren.flower@jenner.ac.uk
WHOLE ORGANISM SUBUNIT VACCINE EPITOPE VACCINE COMPUTATIONAL VACCINOLOGY Vaccines induce protective immunity, an enhanced adaptive immune response to re-infection.
COMPUTATIONAL VACCINOLOGY World class database Antigens, B cell and T cell epitopes Peptide binding, Protein-Protein Interactions Improved Prediction Class I and II T cell epitope prediction B cell epitopes and Antigens Experimental verification and data discovery test prediction and generate new binding data
T-cell, TCR and MHC TCR, MHC and co-receptors on the surface of T-cell and antigen-presenting cell. T-cells have T cell receptors in their membranes that bind to the protein fragments presented by the MHC proteins. T cells recognise the presence of foreign protein and hence pathogenic micro-organisms and then destroy them.
TCR-peptide-MHC complex Peptide MHC binding is just like the binding of drugs to other receptors We can use QSAR and molecular dynamics (MD) simulation to examine, model and predict MHC-peptide interaction
DATA DRIVENMODELLING: QSAR Irini Doytchinova Channa Hattutawagama Valerie Walshe PingPing Guan
QSAR pIC5 pred Y=X*W*(P’*W)-1*C’+F + IC50s pIC50 exp QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP STRUCTURAL DESCRIPTION and BIOLOGICAL RESPONSE PREDICTIVE QSAR MODEL ROBUSTMULTIVARIATE STATISTICS
152 peptides 152 peptides with affinity to with affinity to the HLA-A2.1 the HLA-A2.1 Training set Training set Test set Test set 102 102 50 50 peptides peptides peptides peptides Comparison of CoMFA and CoMSIA for HLA-A*0201 r2pred < 0.5 NC = 6 q2 = 0.480 r2 = 0.911 r2pred = 0.679 NC = 5 q2 = 0.542 r2 = 0.870
Full CoMSIA Analysis of HLA-A*0201 Steric Map Hydrogen Bond Map Electrostatic Map Hydrophobic Map NC = 7 q2 = 0.683 r2 = 0.891 n = 236
P 2 P4 P 6 P 8 H O H O H O H O H O H N N N N N H N N N N O H O H O H O H O P 1 P 3 P 5 P 7 P 9 ADDITIVE METHOD FOR AFFINITY PREDICTION HLA-A*0201: NC = 5 q2 = 0.337 r2 = 0.898 n = 340
STRUCTURE DRIVEN MODELLING: ATOMISTIC MOLECULAR DYNAMIC SIMULATION Shunzhou Wan
PREDICT DYNAMIC PROPERTIES Molecular Dynamics Simulations PREDICT THERMODYNAMICS OF BINDING PREDICT COMPLEX BEHAVIOUR PREDICT PROTEIN - PROTEIN INTERACTIONS Large Molecule Docking DESIGN MUTANTS Point Mutants Chimeras Deletion Mutants Fusion Proteins PREDICT LIGANDS Small Molecule Docking Drug Design ANTIBODY BINDING MHC-peptide BINDING
High Performance Computing& Biomolecular Simulations Simulations of Biomolecular Systems include proteins, nucleic acids, drug-receptor interactions, protein folding, and a few examples of more complex systems, such protein-membrane interactions. Most simulations done on desktop workstations and “small” parallel machines (~32 processors) Long time scales and large systems generally intractable HPC and the GRID allow us, for the first time, to do things properly
Simulated systems are LARGE: 30,000-300,000 atoms Simulation timescales are LONG: In nanoseconds, even microsecond1 Requires high performance computing We use scalable codes LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) & NAMD on large parallel machines (up to 1000+ nodes) Large Scale Molecular Dynamics 1. Duan Y, et al., Science 1998, 282:740-744.
MD scaling performance (LAMMPS) Parallelising the AMBER software scales very poorly in our hands
MHC-peptide complexes HLA-A*0201:MAGE-A4 complex Simulated using AMBER force field in LAMMPS
MHC-peptide complexes: What has been done? a) … 1- 2 domains periodic boundary no constraints Rognan et al. (1992) Proteins 13, 70-85 b) c) 1- 2 domains periodic boundary constraints on backbone Meng et al. (1997) Int. Immunol. 9, 1339-1346 all domains spherical boundary fix all atoms out of sphere constraints on outer buffer region of sphere Michielin et al. (2002) J. Mol. Biol. 324, 547-569
MHC-peptide complexes Can the a3 and b2m domains and/or their movement be neglected in simulations?
MHC-peptide complexes: Simulation models • Many authors1 regards this system as being out of reach of MD simulation • "much too large" • "relevant time scales inaccessible" • But, with scalable codes and tightly coupled massively parallel machines ... Partial model 30,574 atoms No constraints Full model 58,825 atoms No constraints Amber 98 Force Field 1. Nojima et al., Chem Pharm Bull (Tokyo) 2002 50(9), 1209-1214.
MHC-peptide complexes: Simulation models ... for the 58,825 atom model (whole model), we can perform 1 ns simulation in 17 hours' wall clock time on 256 processors of Cray T3E using LAMMPS
MHC-peptide complexes: Results For the partial system, about 300ps were required for equilibration, while the whole system required about 600ps, equilibration here being a function of the size of the system. RMS deviation from x-ray structure versus simulation time (ps). Above: partial MHC-peptide system; Below: whole MHC-peptide system. Solid line: mainchain of protein; Dotted line: mainchain of peptide.
MHC-peptide complexes: Results View of the b-sheets of the average structures from the partial system simulation (blue) and the whole system simulation (yellow), compared with the x-ray structure (red). From top to bottom, the sheets are juxtaposed from the N-terminal to the C-terminal of the peptide. The view is directly onto the peptide-binding side. In the partial system simulation, the middle sheets (4, 5) at the bottom of groove bulge towards the peptide, while 1, 2 and 8 turn aside from it, whereas the whole system simulation does not exhibit these effects.
MHC-peptide complexes: Results RMS deviations of peptide (top) and antigen-binding site of MHC protein (bottom) from x-ray structure. Solid line: whole system simulation; Dashed line: partial system simulation. The loop regions have large deviations from the x-ray structure, while the two long helices and the b-sheets have relatively small deviations.
Peptide Fluctuation vs Thermal B-Factor B FACTOR The larger deviations observed with the peptide in the partial system indicate that the peptide is considerably less tightly bound in the partial system than in the whole system.
MHC-peptide complexes: Conclusions • For 58,825 atoms system, 1 ns simulation can be performed in 17 hours' wall clock time on 256 processors of T3E. • More accurate results are obtained by simulating the whole complex than just a part of it. • The a3 and b2m domains have a significant influence on the structural and dynamical features of the complex, which is very important for determining the binding efficiencies of epitopes. We are now doing TCR-peptide-MHC simulations (~ 100,000 atom model) using NAMD.
WE WANT USE MD TO ADDRESS FUNDAMENTAL PROBLEMS IN IMMUNOLOGY MHCs are polymorphic – there are hundreds of individual alleles with in the human population each with a different peptide binding specificity Use MD to identify binding epitopes and use these to design and develop novel vaccines Also want to use MD to examine more complex systems such as the Immunological Synapse which are not accessible to direct experimental analysis
ACKNOWLEDGEMENTS Bioinformatics Group Irini Doytchinova Shunzhou Wan Helen McSparron Valerie Walshe PingPing Guan Martin Blythe Debra Taylor EJIVR Seph Borrow Outside Peter Coveney (UCL) Funding from EPSRC (RealityGrid, CSAR) Jenner Institute (GSK, BBSRC, MRC, DOH)