Molecular Docking
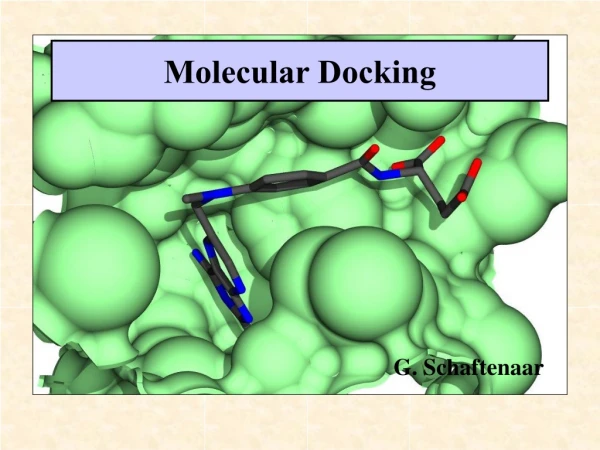
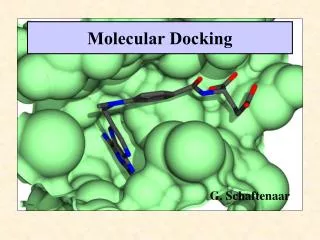
Molecular Docking. G. Schaftenaar. Docking Challenge. Identification of the ligand’s correct binding geometry in the binding site ( Binding Mode ) Observation: Similar ligands can bind at quite different orientations in the active site. Two main tasks of Docking Tools.

Molecular Docking
E N D
Presentation Transcript
Molecular Docking G. Schaftenaar
Docking Challenge • Identification of the ligand’s correct binding geometry in the binding site (Binding Mode) • Observation: • Similar ligands can bind at quite different orientations in the active site.
Two main tasks of Docking Tools • Sampling of conformational (Ligand) space • Scoring protein-ligand complexes
Rigid-body dockingalgorithms • Historically the first approaches. • Protein and ligand fixed. • Search for the relative orientation of the two molecules with lowest energy. • FLOG (Flexible Ligands Oriented on Grid): each ligand represented by up to 25 low energy conformations.
Introducing flexibility:Whole molecule docking • Monte Carlo methods (MC) • Molecular Dynamics (MD) • Simulated Annealing (SA) • Genetic Algorithms (GA) Available in packages: AutoDock (MC,GA,SA) GOLD (GA) Sybyl (MD)
Monte Carlo • Start with configuration A (energy EA) • Make random move to configuration B (energy EB) • Accept move when: EB < EA or if EB > EA except with probability P:
Molecular Dynamics • force-field is used to calculate forces on each atom of the simulated system • following Newton mechanics, calculate accelerations, velocities and new coordinates from the forces. (Force = mass times acceleration) • The atoms are moved slightly with respect to a given time step
Simulated Annealing Finding a global minimium by lowering the temperature during the Monte Carlo/MD simulation
Genetic Algorithms • Ligand translation, rotation and configuration variables constitute the genes • Crossovers mixes ligand variables from parent configurations • Mutations randomly change variables • Natural selection of current generation based on fitness • Energy scoring function determines fitness
Introducing flexibility: Fragment Based Methods • build small molecules inside defined binding sites while maximizing favorable contacts. • De Novo methods construct new molecules in the site. • division into two major groups: • Incremental construction (FlexX, Dock) • Place & join.
Placing Fragments and Rigid Molecules • All rigid-body docking methods have in common that superposition of point sets is a fundamental sub-problem that has to be solved efficiently: • Geometric hashing • Pose clustering • Clique detection
Geometric hashing • originates from computer vision • Given a picture of a scene and a set of objects within the picture, both represented by points in 2d space, the goal is to recognize some of the models in the scene
For each triangle of receptor compute the transformation to each ligand matching triangle. Cluster transformations. Score the results. Pose-Clustering
Clique-Detection • Nodes comprise of matches between protein and ligand • Edges connect distance compatible pairs of nodes • In a clique all pair of nodes are connected
Scoring Functions • Shape & Chemical Complementary Scores • Empirical Scoring • Force Field Scoring • Knowledge-based Scoring • Consensus Scoring
Shape & Chemical Complementary Scores • Divide accessible protein surface into zones: • Hydrophobic • Hydrogen-bond donating • Hydrogen-bond accepting • Do the same for the ligand surface • Find ligand orientation with best complementarity score
Empirical Scoring Scoring parameters fit to reproduce Measured binding affinities (FlexX, LUDI, Hammerhead)
D = D + D ´ G G G N 0 rot rot ( ) å + D D D a G f R , hb - . neutral H bonds ( ) å + D D D a G f R , io - . ionic int ( ) å + D D D a G f R , arom . arom int ( ) å + D D D a G f R , lipo . . lipo cont Empirical scoring Loss of entropy during binding Hydrogen-bonding Ionic interactions Aromatic interactions Hydrophobic interactions
q q j r û ij Force Field Scoring (Dock) é ù A B lig prot å å ú ij ij i = - + E c ê nonbond ú 12 6 r r ê ë i j ij ij • Nonbonding interactions (ligand-protein): • van der Waals • -electrostatics • Amber force field
[ ] ( ) ( ) s s = - b s s P , P exp F , p l ref p l Knowledge-based Scoring Function Free energies of molecular interactions derived from structural information on Protein-ligand complexes contained in PDB Boltzmann-Like Statistics of Interatomic Contacts.
Distribution of interatomic distances is converted into energy functions by inverting Boltzmann’s law. P(N,O) F
s s ij ij bulk bulk Potential of Mean Force (PMF) ( ) æ ö s ij r ( ) ( ) ç ÷ seg = - i F r k T ln f r ç ÷ ij B Vol _ corr è ø ( ) s ij r Number density of atom pairs of type ij at atom pair distance r seg Number density of atom pairs of type ij in reference sphere with radius R
Consensus Scoring Cscore: Integrate multiple scoring functions to produce a consensus score that is more accurate than any single function for predicting binding affinity.
Virtual screening by Docking • Find weak binders in pool of non-binders • Many false positives (96-100%) • Consensus Scoring reduces rate of false positives
Scoring functions are the Achilles’ heel of docking programs. False positives rates can be reduced using several scoring functions in a consensus-scoring strategy Although the reliability of docking methods is not so high, they can provide new suggestions for protein-ligand interactions that otherwise may be overlooked Concluding remarks