Download

1 / 15
650 likes | 1.75k Vues
LA TALASSEMIA. La storia. Nel 1925 negli Stati Uniti d’America i pediatri Cooley e Lee descrissero una malattia, che fu denominata morbo di Cooley, in figli di immigrati italiani e greci. Oggi tale malattia è conosciuta come talassemia maior o anemia mediterranea.

E N D
La storia Nel 1925 negli Stati Uniti d’America i pediatri Cooley e Lee descrissero una malattia, che fu denominata morbo di Cooley, in figli di immigrati italiani e greci. Oggi tale malattia è conosciuta come talassemia maior o anemia mediterranea. In contemporanea in Italia Rietti descrisse una malattia simile, con sintomatologia più lieve, che allora fu chiamata malattia di Rietti Greppi Micheli, e che oggi è conosciuta come talassemia intermedia. In Italia, nel 1943 due ematologi, Ezio Silvestroni ed Ida Bianco, descrissero una malattia ereditaria, individuata in soggetti sani denominata «microcitemia». Silvestroni e Bianco dimostrarono che il morbo di Coooley era una malattia autosomica recessiva che ha origine dalla condizione omozigotica, cioè che il figlio malato nasce solo se entrambi i genitori gli trasmettono l’alterazione microcitemica.
Le cause Con circa 4000 malati (in passato erano più di 8000) e 2.500.000 di portatori sani (eterozigoti) la talassemia (da thalathos, mare e haima, sangue), o anemia mediterranea, è la malattia genetica più diffusa in Italia; è caratterizzata da un difetto nella sintesi dell’emoglobina. L’emoglobina è la proteina, presente nei globuli rossi, deputata al trasporto dell’ossigeno e dell’anidride carbonica. È formata da quattro catene polipeptidiche: 2 alfa e 2 beta. Sequenze anomale di una di queste catene causano i diversi tipi di sindromi talassemiche, in particolare la beta-talassemia e l’alfa-talassemia, così denominate in quanto il difetto è, rispettivamente, a carico della catena beta e della catena alfa. In Italia, ed in tutto il bacino del Mediterraneo, la forma di talassemia più diffusa è la beta-talassemia.
Le emoglobine umane normali Vi sono tre tipi di emoglobina umana normale: l’Hb A, che è propria dell’individuo dopo la nascita, costituisce il 98% di tutta l’emoglobina ed è composta da 4 catene globiniche: 2 α e 2 β; l’Hb A2, che è anch’essa presente solo dopo la nascita, è composta da 2 catene α e 2 δ, e costituisce il restante 2%; l’emoglobina fetale (Hb F) che è propria del periodo della vita intrauterina ed è formata da 2 catene α e 2 γ.
L’emoglobina Hb A L’emoglobina A (Hb A) è costituita da 2 catene alfa e 2 beta, globine rappresentate in colori diversi nella figura. Ognuna delle quattro catene polipeptidiche, per un totale di 600 aminoacidi, è inoltre ripiegata intorno ad un gruppo prostetico, detto eme, contenente ferro. Al ferro del gruppo eme si legano labilmente l’ossigeno e l’anidride carbonica durante gli scambi gassosi.
“I geni” globinici Come per ogni altro carattere ereditario, anche per il carattere “emoglobina”, e più precisamente per ogni tipo di catena globinica (α, β, γ, δ), esiste nel Dna di ciascun individuo una o due coppie di geni, trasmessi uno dal padre e uno dalla madre. L’individuo, a sua volta, li trasmetterà ai propri figli. Questi geni hanno una ben precisa localizzazione in un tratto di DNA dei cromosomi 11 (per la catena globinica β) e 16 ( per la catena globinica α). Ogni gene è composto da 3 esoni, che sono i tratti di DNA tradotti in catena globinica, e 2 introni, che invece non sono tradotti in globina. Cariotipo umano
I geni globinici nelle talassemie I geni globinici hanno dunque il compito di produrre l’emoglobina: possono però essere assenti (delezioni) o avere difetti di vario genere (mutazioni) e quindi non produrre più o produrre solo una piccola quantità di catene globiniche. Una particolare tecnica (lo studio della sintesi globinica in vitro) rivela con esattezza se è ridotta la sintesi delle catene α o β. Nella β-talassemia frequentemente è il braccio corto del cromosoma 11 a presentare una mutazione puntiforme, con sostituzione di una singola base azotata nella tripletta competente: l’Adenina viene erroneamente sostituita dall’Uracile. Come conseguenza durante la sintesi della globina beta l’acido glutammico in posizione 6 è sostituito dalla valina: un solo amminoacido su 300 è nella posizione sbagliata. Nell’α-talassemia i geni coinvolti sono sul cromosoma 16, due geni per ogni cromosoma, per un totale di 4 copie. La gravità della malattia è dovuta a quante di queste quattro copie sono difettose o mancanti. Il quadro che ne deriva è molto articolato. La mancanza di tutte e quattro le copie porta ad una condizione non compatibile con la vita.
La β-thalassemia L’eterozigosi per la β-talassemia configura il quadro della microcitemia. Gli eterozigoti (portatori sani: hanno ereditato un solo allele difettoso) presentano globuli rossi più piccoli del normale e con la membrana leggermente deformata. Lo stato di omozigote, invece, definisce il quadro più grave: la menomata funzione respiratoria dei globuli rossi rende necessarie cure continue, soprattutto trasfusioni di sangue, che devono continuare per tutta la vita. L’unica terapia curativa è il trapianto di midollo osseo (da donatore compatibile), che sostituisce le cellule portatrici del difetto genetico con le cellule del donatore sano. La β-thalassemia omozigote si sviluppa quando sono repressi ambedue i geni, che governano la sintesi delle catene globiniche β e che provengono dai due genitori. Subentra uno squilibrio tra formazione di catene β, che praticamente cessa, e sintesi di catene α, che procede normalmente: di conseguenza la produzione di emoglobina A è fortemente limitata. Ne deriva che: • gli eritroblasti non riescono ad accumulare una quantità di emoglobina sufficiente alla formazione di cellule adeguatamente emoglobinizzate, per cui compare una grave anemia ipocromica; • l’iperabbondanza di catene α, spaiate, ne provoca l’aggregazione sotto forma di corpi Heinz-simili, intraeritrocitari, che danneggiano e deformano tali cellule.
Sangue normale Sangue talassemico Nel sangue di individui normali i globuli rossi appaiono uniformi per dimensioni, forma e colorazione. Nell’individuo affetto da talassemia i globuli rossi appaiono irregolari per forma, dimensione e colorazione. Molti sono solo frammenti (schistociti, freccia rossa). Sono presenti le tipiche cellule a bersaglio.
La trasmissione ereditaria Microcitemia e talassemia si trasmettono come caratteri mendeliani dominanti: passano cioè dai genitori ai figli nel momento del concepimento senza salti di generazioni. Si possono verificare diverse situazioni: • Un microcitemico sposa un soggetto normale: i figli saranno al 50% microcitemici (portatori sani ) e al 50% normali; quindi tutti sani .
La trasmissione ereditaria (segue) • Se invece un microcitemico sposa un altro microcitemico, potranno nascere, oltre a figli sani, sia normali (25%) sia microcitemici (50%), anche figli che avendo ereditato il gene malato da entrambi i genitori saranno malati di anemia mediterranea o talassemia maior (25%):
La diffusione in Italia La microcitemia è diffusa in tutto il mondo con caratteristiche genetiche differenti nelle varie zone. In Italia raggiunge frequenze particolarmente elevate (8 – 15 %) in tutto il Sud, nelle isole e nel delta padano. In tutte le altre zone l’incidenza del portatore sano è intorno al 1,5 – 2 %.
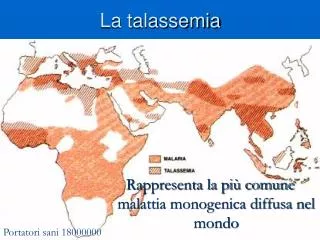
La diffusione nel mondo La microcitemia si presenta con frequenza elevata in tutti i paesi del Mediterraneo (Albania, tutte le nazioni che si affacciano sul mare Adriatico, Grecia, Cipro, Turchia, Marocco, Algeria, Libia, Egitto, il territorio della Palestina, Siria). Si calcola che in questo vasto territorio ci siano circa 180.000.000 di portatori sani e che in assenza di prevenzione ogni anno nascano 70.000 nuovi malati. In Africa (particolarmente fra i tropici), in Sicilia, in Usa nella popolazione di colore, in Brasile e nel Sud est asiatico alla microcitemia è spesso associata una diversa emoglobinopatia (l’anemia falciforme).
Talassemia e malaria Sembra ormai chiaro da diverso tempo che la endemizzazione delle talassemie nelle zone indicate sia collegata alla diffusione analoga della malaria, che ha funzionato come agente selezionatore. È infatti molto stretta la correlazione esistente fra l’incidenza della malaria e quella della β-talassemia. Proprio lo studio dell’incidenza della malaria ha dimostrato che gli individui eterozigoti, affetti quindi da talassemia minor o microcitemia, erano meno sensibili alla malaria degli omozigoti normali. Questo si spiega col fatto che l’agente della malaria, Plasmodium falciparum, si sviluppa all’interno dei normali globuli rossi dell’ospite che infetta, e li distrugge. L’individuo eterozigote per la talassemia possiede una quota di globuli rossi alterati con una vita più breve del normale, risulta quindi più difficile il completamento del ciclo riproduttivo del plasmodio. Nel tempo questo ha permesso agli eterozigoti di riprodursi con maggior successo dei soggetti che si ammalano di malaria, comportando la trasmissione del gene alterato alle generazioni successive. Sebbene quindi la selezione agisca contro gli omozigoti malati eliminando i geni dannosi, il vantaggio dell’eterozigote ha mantenuto l’allele recessivo nella popolazione.
FontiAlberghina – Tonini “Logica ed applicazioni della biologia” Arnoldo Mondadori ScuolaKapf – Jandl “Sangue, testo edAtlante di ematologia” Verduci EditoreRosa – Simonelli – Spanò “Moduli di Biologia” Garzanti Scuola“Microcitemie ed anemia mediterranea” Regione Lazio – Associazione Nazionale per la lotta contro la microcitemia in Italiahttp://www.blod.info/it/domande-frequenti.asphttp://it.wikipedia.org/wiki/Pagina_principale