Download
1 / 1
10 likes | 104 Vues
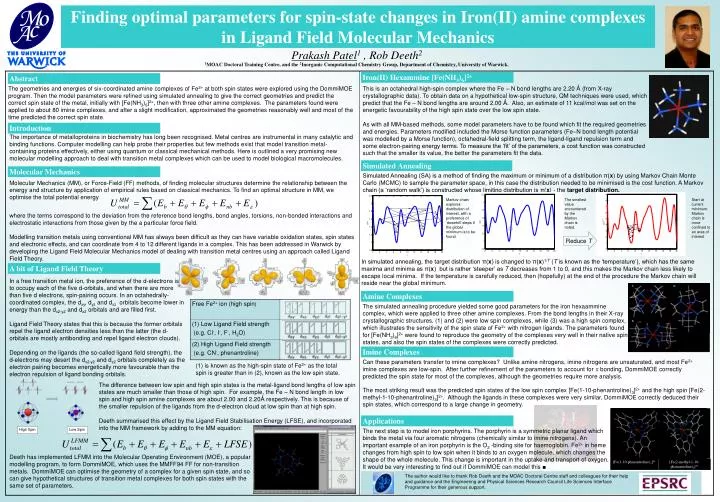
Finding optimal parameters for spin-state changes in Iron(II) amine complexes in Ligand Field Molecular Mechanics. Prakash Patel 1 , Rob Deeth 2. 1 MOAC Doctoral Training Centre, and the 2 Inorganic Computational Chemistry Group, Department of Chemistry, University of Warwick. 1. 2.

E N D
Finding optimal parameters for spin-state changes in Iron(II) amine complexes in Ligand Field Molecular Mechanics Prakash Patel1 , Rob Deeth2 1MOAC Doctoral Training Centre, and the 2Inorganic Computational Chemistry Group, Department of Chemistry, University of Warwick. 1. 2. High Spin Low Spin 3. Reduce T Free Fe2+ ion (high spin) (1) Low Ligand Field strength (e.g. Cl-, I-, F-, H2O) (2) High Ligand Field strength (e.g. CN-, phenantroline) [Fe(1-10-phenantroline)3]2+ [Fe(2-methyl-1-10-phenantroline)3]2+ The author would like to thank Rob Deeth and the MOAC Doctoral Centre staff and colleagues for their help and guidance and the Engineering and Physical Sciences Research Council Life Sciences Interface Programme for their generous support. Iron(II) Hexammine [Fe(NH3)6]2+ Abstract The geometries and energies of six-coordinated amine complexes of Fe2+ at both spin states were explored using the DommiMOE program. Then the model parameters were refined using simulated annealing to give the correct geometries and predict the correct spin state of the metal, initially with [Fe(NH3)6]2+, then with three other amine complexes. The parameters found were applied to about 80 imine complexes, and after a slight modification, approximated the geometries reasonably well and most of the time predicted the correct spin state. This is an octahedral high-spin complex where the Fe – N bond lengths are 2.20 Ǻ (from X-ray crystallographic data). To obtain data on a hypothetical low-spin structure, QM techniques were used, which predict that the Fe – N bond lengths are around 2.00 Ǻ. Also, an estimate of 11 kcal/mol was set on the energetic favourability of the high spin state over the low spin state. As with all MM-based methods, some model parameters have to be found which fit the required geometries and energies. Parameters modified included the Morse function parameters (Fe–N bond length potential was modelled by a Morse function), octahedral-field splitting term, the ligand-ligand repulsion term and some electron-pairing energy terms. To measure the ‘fit’ of the parameters, a cost function was constructed such that the smaller its value, the better the parameters fit the data. Introduction The importance of metalloproteins in biochemistry has long been recognised. Metal centres are instrumental in many catalytic and binding functions. Computer modelling can help probe their properties but few methods exist that model transition metal-containing proteins effectively, either using quantum or classical mechanical methods. Here is outlined a very promising new molecular modelling approach to deal with transition metal complexes which can be used to model biological macromolecules. Simulated Annealing Molecular Mechanics Simulated Annealing (SA) is a method of finding the maximum or minimum of a distribution π(x) by using Markov Chain Monte Carlo (MCMC) to sample the parameter space, in this case the distribution needed to be minimised is the cost function. A Markov chain (a ‘random walk’) is constructed whose limiting distribution is π(x) - the target distribution. Molecular Mechanics (MM), or Force-Field (FF) methods, of finding molecular structures determine the relationship between the energy and structure by application of empirical rules based on classical mechanics. To find an optimal structure in MM, we optimise the total potential energy Markov chain explores distribution of interest, with a preference of ‘downhill’ steps if the global minimum is to be found. The smallest value encountered by the Markov chain is noted. Start at current minimum. Markov chain is more confined to an area of interest where the terms correspond to the deviation from the reference bond lengths, bond angles, torsions, non-bonded interactions and electrostatic interactions from those given by the a particular force field. Modelling transition metals using conventional MM has always been difficult as they can have variable oxidation states, spin states and electronic effects, and can coordinate from 4 to 12 different ligands in a complex. This has been addressed in Warwick by developing the Ligand Field Molecular Mechanics model of dealing with transition metal centres using an approach called Ligand Field Theory. In simulated annealing, the target distribution π(x) is changed to π(x)1/T (T is known as the ‘temperature’), which has the same maxima and minima as π(x) but is rather ‘steeper’ as T decreases from 1 to 0, and this makes the Markov chain less likely to escape local minima. If the temperature is carefully reduced, then (hopefully) at the end of the procedure the Markov chain will reside near the global minimum. A bit of Ligand Field Theory In a free transition metal ion, the preference of the d-electrons is to occupy each of the five d-orbitals, and when there are more than five d electrons, spin-pairing occurs. In an octahedrally-coordinated complex, the dxy, dyz and dxz orbitals become lower in energy than the dx2-y2 and dz2 orbitals and are filled first. Ligand Field Theory states that this is because the former orbitals repel the ligand electron densities less than the latter (the d-orbitals are mostly antibonding and repel ligand electron clouds). Depending on the ligands (the so-called ligand field strength), the d-electrons may desert the dx2-y2 and dz2 orbitals completely as the electron pairing becomes energetically more favourable than the electron repulsion of ligand bonding orbitals. Amine Complexes The simulated annealing procedure yielded some good parameters for the iron hexaammine complex, which were applied to three other amine complexes. From the bond lengths in their X-ray crystallographic structures, (1) and (2) were low spin complexes, while (3) was a high spin complex, which illustrates the sensitivity of the spin state of Fe2+ with nitrogen ligands. The parameters found for [Fe(NH3)6]2+ were found to reproduce the geometry of the complexes very well in their native spin states, and also the spin states of the complexes were correctly predicted. Imine Complexes Can these parameters transfer to imine complexes? Unlike amine nitrogens, imine nitrogens are unsaturated, and most Fe2+ imine complexes are low-spin. After further refinement of the parameters to account for p bonding, DommiMOE correctly predicted the spin state for most of the complexes, although the geometries require more analysis. The most striking result was the predicted spin states of the low spin complex [Fe(1-10-phenantroline)3]2+ and the high spin [Fe(2-methyl-1-10-phenantroline)3]2+. Although the ligands in these complexes were very similar, DommiMOE correctly deduced their spin states, which correspond to a large change in geometry. (1) is known as the high-spin state of Fe2+ as the total spin is greater than in (2), known as the low spin state. The difference between low spin and high spin states is the metal-ligand bond lengths of low spin states are much smaller than those of high spin. For example, the Fe – N bond length in low spin and high spin amine complexes are about 2.00 and 2.20Ǻ respectively. This is because of the smaller repulsion of the ligands from the d-electron cloud at low spin than at high spin. Deeth summarised this effect by the Ligand Field Stabilisation Energy (LFSE), and incorporated into the MM framework by adding to the MM equation: Applications The next step is to model iron porphyrins. The porphyrin is a symmetric planar ligand which binds the metal via four aromatic nitrogens (chemically similar to imine nitrogens). An important example of an iron porphyrin is the O2 -binding site for haemoglobin. Fe2+ in heme changes from high spin to low spin when it binds to an oxygen molecule, which changes the shape of the whole molecule. This change is important in the uptake and transport of oxygen. It would be very interesting to find out if DommiMOE can model this ■ Deeth has implemented LFMM into the Molecular Operating Environment (MOE), a popular modelling program, to form DommiMOE, which uses the MMFF94 FF for non-transition metals. DommiMOE can optimise the geometry of a complex for a given spin state, and so can give hypothetical structures of transition metal complexes for both spin states with the same set of parameters.