Download
1 / 1
10 likes | 186 Vues
Xander : Gene-Targeted Metagenomic Assembly. Jordan A. Fish 1 , Qiong Wang 1 , Yanni Sun 2 , C. Titus Brown 2,3 , James M. Tiedje 1,3 and James R. Cole 1. 1 Center for Microbial Ecology; 2 Department of Computer Science and Engineering; 3 Department of Microbiology and Molecular Genetics

E N D
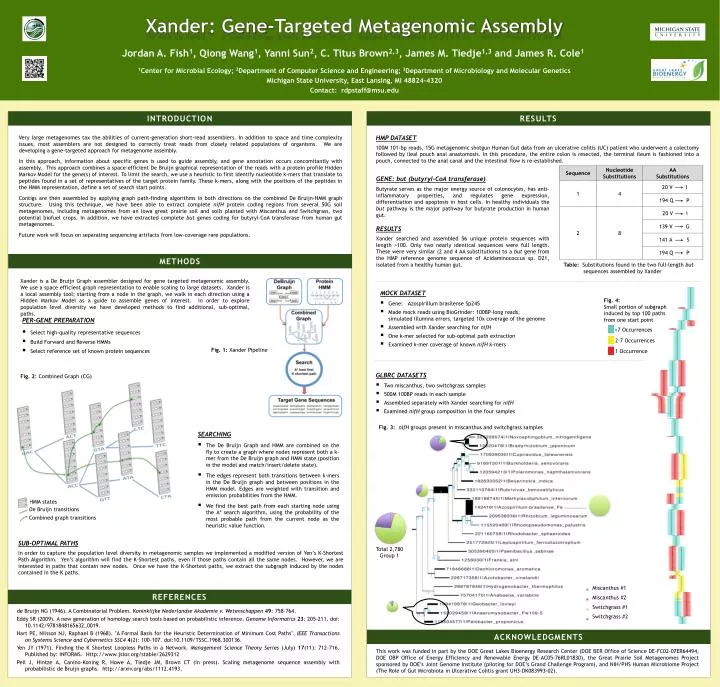
Xander: Gene-Targeted Metagenomic Assembly Jordan A. Fish1, Qiong Wang1, Yanni Sun2, C. Titus Brown2,3, James M. Tiedje1,3 and James R. Cole1 1Center for Microbial Ecology; 2Department of Computer Science and Engineering; 3Department of Microbiology and Molecular Genetics Michigan State University, East Lansing, MI 48824-4320 Contact: rdpstaff@msu.edu INTRODUCTION RESULTS Very large metagenomes tax the abilities of current-generation short-read assemblers. In addition to space and time complexity issues, most assemblers are not designed to correctly treat reads from closely related populations of organisms. We are developing a gene-targeted approach for metagenome assembly. In this approach, information about specific genes is used to guide assembly, and gene annotation occurs concomitantly with assembly. This approach combines a space-efficient De Bruijn graphical representation of the reads with a protein profile Hidden Markov Model for the gene(s) of interest. To limit the search, we use a heuristic to first identify nucleotide k-mers that translate to peptides found in a set of representatives of the target protein family. These k-mers, along with the positions of the peptides in the HMM representation, define a set of search start points. Contigs are then assembled by applying graph path-finding algorithms in both directions on the combined De Bruijn-HMM graph structure. Using this technique, we have been able to extract complete nifH protein coding regions from several 50G soil metagenomes, including metagenomes from an Iowa great prairie soil and soils planted with Miscanthus and Switchgrass, two potential biofuel crops. In addition, we have extracted complete but genes coding for butyryl-CoA transferase from human gut metagenomes. Future work will focus on separating sequencing artifacts from low-coverage rare populations. HMP DATASET 100M 101-bp reads, 15G metagenomic shotgun Human Gut data from an ulcerative colitis (UC) patient who underwent a colectomy followed by ileal pouch anal anastomosis. In this procedure, the entire colon is resected, the terminal ileum is fashioned into a pouch, connected to the anal canal and the intestinal flow is re-established. GENE: but (butyryl-CoA transferase) Butyrate serves as the major energy source of colonocytes, has anti-inflammatory properties, and regulates gene expression, differentiation and apoptosis in host cells. In healthy individuals the but pathway is the major pathway for butyrate production in human gut. RESULTS Xander searched and assembled 56 unique protein sequences with length >100. Only two nearly identical sequences were full length. These were very similar (2 and 4 AA substitutions) to a butgene from the HMP reference genome sequence of Acidaminococcus sp. D21, isolated from a healthy human gut. METHODS Table: Substitutions found in the two full-length but sequences assembled by Xander Xander is a De Bruijn Graph assembler designed for gene targeted metagenomic assembly. We use a space efficient graph representation to enable scaling to large datasets. Xander is a local assembly tool; starting from a node in the graph, we walk in each direction using a Hidden Markov Model as a guide to assemble genes of interest. In order to explore population level diversity we have developed methods to find additional, sub-optimal, paths. • MOCK DATASET • Gene: Azospirillumbrasilense Sp245 • Made mock reads using BioGrinder: 100BP-long reads, simulated Illumina errors, targeted 10x coverage of the genome • Assembled with Xander searching for nifH • One k-mer selected for sub-optimal path extraction • Examined k-mer coverage of known nifHk-mers Fig. 4: Small portion of subgraph induced by top 100 paths from one start point • PER-GENE PREPARATION • Select high-quality representative sequences • Build Forward and Reverse HMMs • Select reference set of known protein sequences >7 Occurrences 2-7 Occurrences Fig. 1: Xander Pipeline 1 Occurrence • GLBRC DATASETS • Two miscanthus, two switchgrasssamples • 500M 100BP reads in each sample • Assembled separately with Xander searching for nifH • Examined nifH group composition in the four samples Fig. 2: Combined Graph (CG) Fig. 3: nifHgroups present in miscanthus and switchgrass samples • SEARCHING • The De Bruijn Graph and HMM are combined on the fly to create a graph where nodes represent both a k-mer from the De Bruijn graph and HMM state (position in the model and match/insert/delete state). • The edges represent both transitions between k-mersin the De Bruijngraph and between positions in the HMM model. Edges are weighted with transition and emission probabilities from the HMM. • We find the best path from each starting node using the A* search algorithm, using the probability of the most probable path from the current node as the heuristic value function. HMM states De Bruijn transitions Combined graph transitions SUB-OPTIMAL PATHS In order to capture the population level diversity in metagenomic samples we implemented a modified version of Yen’s K-Shortest Path Algorithm. Yen’s algorithm will find the K-Shortest paths, even if those paths contain all the same nodes. However, we are interested in paths that contain new nodes. Once we have the K-Shortest paths, we extract the subgraph induced by the nodes contained in the K paths. Total 2,780Group 1 Miscanthus #1 Miscanthus #2 Switchgrass #1 Switchgrass #2 REFERENCES de Bruijn NG (1946). A CombinatorialProblem. Koninklijke Nederlandse Akademie v. Wetenschappen49: 758–764. Eddy SR (2009). A new generation of homology search tools based on probabilistic inference. Genome Informatics 23: 205-211. doi: 10.1142/9781848165632_0019. Hart PE, Nilsson NJ, Raphael B (1968). "A Formal Basis for the Heuristic Determination of Minimum Cost Paths". IEEE Transactions on Systems Science and Cybernetics SSC44(2): 100–107. doi:10.1109/TSSC.1968.300136. Yen JY (1971). Finding the K Shortest Loopless Paths in a Network. Management Science Theory Series (July)17(11): 712-716. Published by: INFORMS. Http://www.jstor.org/stable/2629312 Pell J, Hintze A, Canino-Koning R, Howe A, Tiedje JM, Brown CT (in press). Scaling metagenome sequence assembly with probabilistic de Bruijn graphs. http://arxiv.org/abs/1112.4193. ACKNOWLEDGMENTS This work was funded in part by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494, DOE OBP Office of Energy Efficiency and Renewable Energy DE-AC05-76RL01830), the Great Prairie Soil Metagenomes Project sponsored by DOE’s Joint Genome Institute (piloting for DOE’s Grand Challenge Program), and NIH/PHS Human Microbiome Project (The Role of Gut Microbiota in Ulcerative Colitis grant UH3-DK083993-02).