Download

1 / 50
800 likes | 2.41k Vues
Motor Neuron Diseases. Motor Neuron Diseases group of diseases which include progressive degeneration and loss of motor neurons with or without similar lesion of the motor nuclei of the brain replacement of lost cells with gliosis

E N D
Motor Neuron Diseases • group of diseases which include progressive degeneration and loss of motor neurons with or without similar lesion of the motor nuclei of the brain • replacement of lost cells with gliosis • “Motor Neuron Disease” = ALS (Charcot’s Disease, Lou Gehrig’s Disease) • LMN - limbs (PMA), bulbar (progressive bulbar palsy) • UMN – limbs (PLS), bulbar (progressive pseudobulbar palsy)
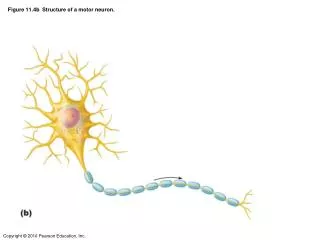
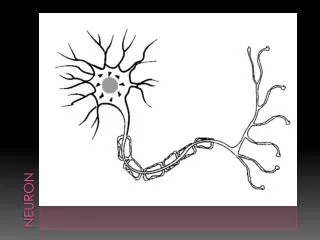
Upper motor neuron Lower motor neuron Diagnostic Triad: ALS Progression
ALS Demographics • Incidence 2 per 100,000 • Male slightly > Female • Peak age of onset: 6th decade (range 20 to 90) • No racial predilection • 95% sporadic • 5% AD (FALS)
ALS Diagnosis: Upper MotorNeuron Symptoms • Loss of dexterity • Slowed movements • Loss of muscle strength • Stiffness • Emotional lability
ALS Diagnosis: Upper Motor Neuron Signs Bulbar Jaw jerk Snout Palmomental Pseudobulbar palsy/ affect Glabellar Cervical Pathologic DTRs Hoffmans Spasticity Thoracic Loss of abdominal reflexes LumbosacralPathologic DTRs, Extensor plantar signs, Spasticity
ALS Diagnosis: Lower Motor Neuron Symptoms • Loss of muscle strength • Atrophy • Fasciculations • Muscle cramps
ALS: Inconsistent Clinical Features • Sensory dysfunction • Bladder and bowel sphincter dysfunction • Autonomic nervous system dysfunction • Visual pathway abnormalities • Movement disorders • Cognitive abnormalities • Bedsores
Pathology • Precentral gyrus atrophy • Sparing of nucleus of Onuf • Neuronal loss of cranial nuclei • Degeneration of corticospinal tract • Chromatin dissolution (chromatolysis), atrophy, shrinkage, cell loss, gliosis
Pathology • Bunina’s bodies – intracytoplasmic, easinophilic dense granular • Hirano’s bodies – rod shaped, contain parallel filaments • Lewy bodies • Neuritic plaques • Neurofibrillary tangles
Familial ALS • AD inheritance, variable penetrance • Male = Female • Higher incidence of cognitive changes • Chorea • Younger onset • Reported spongiform changes, plaques, tangles • 15 year survival • One type maps to chromosome 2 • 20 % are SOD
ALS: Differential Diagnosis • Toxins (lead, mercury, ?aluminum) • Metabolic (hyperthyroidism, hyperparathyroidism, hypoglycemia) • Enzyme deficiency (Hexosaminidase A) • Paraneoplastic (lymphoma, small cell lung) • Cervical spondylosis
ALS: Differential Diagnosis • Immunologic (paraproteinemia) • Multi-system degeneration (Creutzfeldt-Jacob, ALS-PD-Dementia, Spinocerebellar Degeneration) • Viral (Post-polio) • Bacterial (Lyme disease) • Vitamin B12 deficiency
ALS: Laboratory Studies • CK levels are typically normal but may be increased 2-3x normal in almost half of patients.* • CSF may show mild protein elevation (less than 100mg/dl).* • All other laboratory studies should be normal.
ALS: Electrodiagnostic Testing • Normal SNAPs • CMAPs may be normal or show decreased amplitude* • NCV rarely < 80% LLN • DL rarely > 1.5x normal • F response rarely > 1.3x normal • Fibrillations/fasciculations in 2 muscles in 3 extremities (head and paraspinals count as an extremity)*
ALS: Prognosis • Prognosis • 50% dead in 3 years • 20% live 5 years • 10% live 10 years • Worse prognosis if: • Bulbar onset • Simultaneous arm/leg onset • Older age at diagnosis (onset < 40: 8.2 yr duration, onset 61-70: 2.6 yr duration)
Primary Lateral Sclerosis • Upper motor neuron syndrome • Rare disorder (2% of MND cases) with survival ranging between years - decades • Weakness is typically distal, asymmetrical • Patients present with slowly progressive spastic paralysis/bulbar palsy • EMG should not reveal evidence of active or chronic denervation
Primary Lateral Sclerosis • Patients may develop clinical LMN abnormalities over the course of their disease. • Frequently, patients may have subtle evidence of active or chronic denervation on EMG (rare fibs/decreased recruitment), and/or muscle biopsy at diagnosis
Progressive Muscular Atrophy • Lower motor neuron syndrome • Literature suggests 8-10% of patients with MND • Much better prognosis than ALS (mean duration 3-14 years) • Bulbar involvement is rare • Weakness is typically distal, asymmetrical
Multi-focal motor neuropathy Mononeuropathy multiplex CIDP Polyneuropathy/ radiculopathy Plexopathy Kennedy’s Hexosaminidase A deficiency Spinal muscular atrophy Post-polio syndrome Polymyositis Inclusion body myositis LMN onset ALS PMA Lower Motor Neuron Syndromes
Progressive Muscular Atrophy • The majority of patients presenting with PMA eventually develop clinical UMN signs. • Post-mortem examinations of PMA patients frequently show pathologic evidence of UMN degeneration. • In some FALS families, the same gene mutation causes the phenotypes of PMA and ALS in different individuals.
Spinobulbar Muscular Atrophy • Originally reported by Kennedy in 1966 – 11 males in 2 families • Age of onset • Usually begins in 3rd or 4th decade • Genetics • Most common form of adult onset SMA • X-linked recessive • >40 CAG repeats in the androgen receptor gene • Number of repeats correlates with age of onset
Spinobulbar Muscular Atrophy • Lower motor neuron syndrome with limb-girdle distribution of weakness/bulbar palsy* • Facial or perioral fasciculations (90%) • Tongue atrophy with longitudinal midline furrowing • Prominent muscle cramps • Generalized fasciculations and atrophy • Rarely causes respiratory muscle weakness
Spinobulbar Muscular Atrophy • Reflexes are decreased or absent • Cognitive impairment may occur • Hand tremor • Sensory exam may be normal or minimally abnormal
Spinobulbar Muscular Atrophy:Systemic Manifestations • Gynecomastia (60-90%)* • Testicular atrophy (40%) • Feminization • Impotence* • Infertility • Diabetes (10-20%)
Spinobulbar Muscular Atrophy:Laboratory Studies • Markedly abnormal sensory NCS • Sural nerve bx: significant loss of myelinated fibers* • Elevated CK (may be 10x normal) • Abnormal sex hormone levels (androgen nl or decreased; estrogen may be elevated, FSH/LH may be mildly elevated)* • Increased expansion of CAG repeats in the androgen receptor gene*
Conclusions • Although some patients with MND variants evolve into “classic” ALS over time, others continue to show restricted clinical features even late in the course of their disease. • In daily clinical practice, precise definitions may not be crucial but recognition of the “variants” is important since each has a different course and prognosis. • The “treatment cocktail” should be the same until we learn more about pathogenesis.
Treatment Issues to Consider • Symptom management • Nutritional management • Respiratory management • Palliative care • Therapies to slow disease progression
Dysarthria Dysphagia Sialorrhea Emotional lability Depression Weight Loss Bladder urgency Sleep dysfunction Constipation Edema Pain Spasticity Cramps Weight loss Fatigue Weakness Symptoms Associated with Motor Neuron Disease
Sialorrhea • Symptoms result from inability to clear oropharyngeal secretions • Common pharmacologic treatments: • Glycopyrrolate (Robinul) 1-2 mg q 4h • Amitriptyline (Elavil) 25-100 mg qhs • Hyoscyamine sulfate (Levsin) 1-2 tsp q 4h • Transdermal scopolamine • Suction machines
Management of Emotional Lability • Common pharmacologic treatments: • Amitriptyline (Elavil) 25-150 mg qhs* • SSRIs • Common nonpharmacologic treatments: • Counseling/support groups
Spasticity • Common pharmacologic treatments*: • Baclofen (Lioresal) 10-40 mg TID-QID • Dantrolene sodium (Dantrium) 25 mg qd - QID • Tizanidine HCL (Zanaflex) 12-36 mg TID • Diazepam (Valium) 2-5mg TID • Botox ? • Common nonpharmacologic treatments: • Physical therapy • Occupational therapy
Cane Roll-aided walker AFOs Wheelchair Hoyer lift Cervical collar Hospital bed Ramps Built-up utensils Velcro fasteners Raised toilet seat Shower chair Resting hand splints Grab bars Management of Weakness:Assistive Devices
Management of Dysphagia: Consideration for PEG • Consider • Significant weight loss • Inadequate fluid or caloric intake • Difficulty swallowing medications • Frequent choking during meals • Prolonged meal times • FVC < 50% • Aspiration pneumonia* • Does not prolong survival • Malnutrition independent risk factor for worse prognosis
Respiratory Insufficiency: Early Symptoms • Dyspnea on exertion • Supine dyspnea • Marked fatigue • Excessive daytime somnolence • Frequent nocturnal arousals • Vivid dreams • Morning headaches
Management of Respiratory Muscle Weakness • Consider initiation of support when: • Symptoms of nocturnal hypoventilation • FVC <50% of predicted • MIP < -60 cm H2O • Evidence of significant O2 desaturations • May prolong time to death/trach in longitudinal studies
Pathogenesis • Nucleic acid metabolism – decreased nucleolus staining, reduced mRNA/rRNA content • Glutamate – activation NMDA type receptor, Cainflux, free radical production (NO/ROS/protein misfolding by endoplasmic reticulum) • Increased in CSF and plasma • Decreased in brain and spinal cord • Decreased active transport of glutamate into synaptosomes • Loss of glial glutamate transporters
Pathogenesis • Loss of muscarinic cholinergic repectors of anterior horns • Decreased choline acetyltransferase in spinal cord • Decreased glycine and BZD receptors • Immunology • CSF IgG ? Elevated in spinal cord • C3, C4 deposits in spinal cord • Reported abnormal glycolipid antibodies in serum • Elevated antibodies to voltage gated calcium channels – disturbance of calcium homeostasis (binding proteins parvalbumin/calbindinD28)
Pathogenesis • Viral? – amantadine not effective • SOD1 – loss of function mutation? • 20% of FALS • Free radical toxicity • Chromosome 21 • Cytosolic enzyme • Transgenic mouse model
Pathogenesis • Heat shock proteins – chaperones, influence shape, shuttle proteins • Apoptosis – programmed cell death • CNS glial cells – retain some reproductive capacity • Microglial – specialized macrophages • Macroglia – astrocytes, oligodendrocytes, ependymal cells, radial glial (neurogenesis/migration)
Treatment • Riluzole • IGF-1 - growth factor • Ceftriaxone – glutamate transporter • Co-Q10 • Statins • Memantine with riluzole
Treatment • Tamoxifen with riluzole • Celebrex • Thalidomide - TNF alpha • Buspirone – neurotrophic effect • Stem cell*
Western Pacific ALS • ALS-PD-Dementia Guam, West New Guinea, Honshu Island • Earlier onset • UMN precedes LMN features • Bulbar weakness more common
Hexosaminidase A Deficiency • AR • Onset childhood • SMA-like picture • Mild dementia, neuropathy, ataxia, psychosis • Atrophy on imaging (cerebellum)