Download

1 / 15
210 likes | 660 Vues
Steps in a genome sequencing project. Funding and sequencing strategy source of funding identified / community drive development of sequencing strategy random shotgun (chromosome & whole genome)

E N D
Steps in a genome sequencing project Funding and sequencing strategy • source of funding identified / community drive • development of sequencing strategy • random shotgun (chromosome & whole genome) sheared gDNA libraries, physical maps not necessary, fast, whole genome coverage produced quickly, assembly may be problematic • clone-by-clone (map-as-you-go) BAC, YAC, cosmid libraries & physical maps, slower, data produced less quickly from isolated regions • procurement of DNA: library construction, test sequencing, analysis of data • large-scale sequencing of libraries • Assembly and data release • for shotgun projects: at 3 X: first assembly, release of genome data • at 5-6 X: ~97% genes sequenced • at 8-10 X coverage, final assembly • for clone-by-clone: sequence of clones released as completed • Closure • gap closure, repeat resolution, identification of mis-assemblies: time-consuming, expensive • comparison to physical/genetic/optical maps • Gene finding and annotation • train gene finding algorithms and predict gene models • genome annotation: auto-annotation vs manual annotation • genome analysis, comparative genomics, publication, final data release to GenBank
Sequencing strategies for long DNA We can’t directly sequence long DNA (yet), but we can assemble the master sequence from smaller pieces.
Shotgun Library Construction & Sequencing • Concept: • Shred long DNA into lots of random short fragments • Sequence both ends of the fragments • Reassemble the original DNA from overlapping sequences of the fragments • SOUNDS EASY!
Methods: • sonication • syringe • nebulization • NOT RESTRICTION ENZYMES
Size-selected shotgun fragment Libraries • Small insert library provides most of the sequence coverage (contigs) • Large insert libraries help order the contigs (and scaffolds)
5’ end read Mate pair (~1kb between) 3’ end read 5’ end read Mate pair (~9kb between) 3’ end read
Assembly of contigs from mate pairs • must have high-quality (well-trimmed) input DNA, to reduce false overlaps • reads must be mostly mate pairs (<25% single reads) • library insert size variance must be kept low (<10%) for accurate prediction of distance between mate-pairs sequences
Scaffolds, or ‘Why we sequence mate pairs from longer fragments’ low-complexity/repetitive Knowing the sizes of inserts can tell us roughly what we don’t we don’t know (sometimes).
Two ways of thinking about: COVERAGE What does “8X coverage” mean?? • - The average number of times any given base in the genome was sequenced (in this case, each base was read 8 times on average. Of course a particular base may have been read more or less than 8 times.) • also • The amount of sequence that was obtained, relative to the length of the whole genome (in this case, the aggregate length of all reads was 8 times the genome length) • Lander & Waterman (1988) determined that for an ideal genome project (no ‘difficult’ regions) 8X-10X coverage is sufficient to confidently complete the genome.
NO EUKARYOTIC GENOME IS THAT WELL-BEHAVED So even with 8X shotgun coverage there’s likely at least ~1% of the genome remaining to be finished, by more laborious and expensive means (The human genome…are we there yet??) Some genomes are relatively well-behaved: nearly all sequence reads were assembled into contigsscaffoldschromosomes, with relatively few or no gaps remaining (e.g., Plasmodium falciparum) Some genomes are verybadly behaved and far from finished; reads may remain unassigned to contigs, much less scaffolds, much less chromosomes. There are lots of gaps (Ns) and lots of repeats. E.g., Trichomonas vaginalis genome: huge, highly repetitive, AT-rich; low-quality seq was allowed in to increase coverage/gene calls in ‘difficult’ regions..
Finishing • Closure of gaps between contigs/scaffolds • Correction of misassemblies • resequencing of low-coverage/low-quality regions This is usually the most time-consuming part of the project. Repeat/low complexity regions can be hard to sequence and hard to know where to ‘put’ in the final assembly.
Sequence hierarchy genome (all chromosomes) Chromosome (one or more scaffolds..ultimately one contig!) ordered sets w/gaps Scaffold (two or more contigs) ordered sets w/gaps, size estimated Not biological entities contig overlapping, ordered sets, no gaps reads (mate-pair & single)
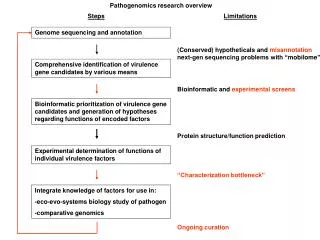
Post-sequencing steps Automated • gene calling (setting boundaries) • Annotation (guessing function) Manual • refining gene models • correcting annotation • should be an ONGOING process…wish it was
OTHER STUFF (demonstrated on the websites) Adding columns Sorting (some are presorted) Gaps: more than one N (within scaffold, gap between scaffold), vs ambiguities (contig) (see P.falc) Chromosome as one giant contig…or one giant scaffold