Genome sequencing and assembling
140 likes | 458 Vues
Genome sequencing and assembling. Chitta Baral. Basic ideas and limitations. Current lab techniques can sequence small (say 700 base pairs) DNA pieces. Use restriction enzymes to cut DNA pieces Sort pieces of different sizes using gel electrophoresis and use the sorting to read them

Genome sequencing and assembling
E N D
Presentation Transcript
Genome sequencing and assembling Chitta Baral
Basic ideas and limitations • Current lab techniques can sequence small (say 700 base pairs) DNA pieces. • Use restriction enzymes to cut DNA pieces • Sort pieces of different sizes using gel electrophoresis and use the sorting to read them • Mapping and Walking • Sequence one piece, get 700 letters, make a primer that allowed you to read the next 700, and work sequentially down the clone • Estimate for human genome sequencing using this method: 100 years • Shotgun sequencing (introduced by Sanger et al. 1977) for sequencing genomes • Obtain random sequence reads from a genome • Assemble them into contigs on the basis of sequence overlaps • Straightforward for simple genomes (with no or few repeat sequences) • Merge reads containing overlapping sequence • Shotgun sequencing is more challenging for complex (repeat-rich) genomes: two approaches
Shotgun sequencing – 2 approaches • Hierarchical shotgun approach • Generating an overlapping set of intermediate-sized (e.g. bacterial artificial chromosomes with 200 KB inserts) clones, and keeping a map of that (it took 2 yrs for mapping e-coli) • Subjecting each of these clones to shotgun sequencing, and using the map to get the whole sequence. • Used in S. cerevisiae (yeast), C. elegans (nematode), A. thaliana (mustard weed) and by the International Human Genome Sequencing Consortium (started in 1990, draft made available in 2000) • Whole-genome shotgun (WGS) approach • Generating sequence reads directly from a whole-genome library • Using computational techniques to reassemble in one step. • Used for Drosophila melanogaster (fruit fly) and by Celera Genomics (formed 1998) for human genome.
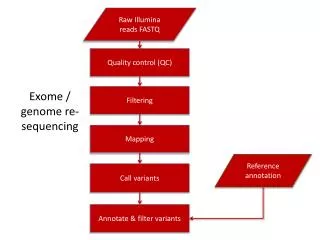
Sequencing small DNA pieces • Use DNA cloning or PCR to make multiple copies. • Put in 4 testtubes marked G, A, T and C • In testtube G use restriction enzymes that cuts at G. • Do the above step for the other testubes. • Use gel electrophoresis separately for the content in each testtube. • The data results in the table on the left. • Reading the table we get G has lengths 1, 7, 12, 13, 19; A has lengths 2, 6, 8, 11, 14,15,16; T has length 4, 5, 9, 18 and C has length 3, 10, 17. • This gives us the sequence.
The ARACHNE WGS assembler: outline of assembly algorithm • Input data: • Paired end reads obtained by sequencing both ends of a plasmid of known insert size. • Assumes each base in each read has an associated quality score (say one obtained by PHRED program) • Quality score q corresponds to the probability 10-q/10 that the base is incorrect (40 corresponds to 99.99% accuracy) • Initial step: eliminates terminal regions whose quality is low. • Eliminates reads containing very little high-quality sequence • Eliminates known vector sequences and known contaminants (eg. Sequence from the bacterial host or cloning vector)
Cont. • Overlap detection and alignment • Create a sorted table of each k-letter subword (k-mer) together with its source (which read) and its position within the read. • Exclude k-mers that occur with extremely high frequency • corresponds to highly repeated sequences; • used to increase the efficiency of the overlap detection process • Identify all instances of read pairs that share one or more overlapping k-mer, and a 3 step process (similar to FASTA) to align the reads effciently • (i) Merge overlapping shared k-mers, (ii) Extend the shared k-mers to alignments, (iii) Refine the alignment by dynamic programming. • Some valid alignments may be missed and some invalid ones may result.
ARACHNE: Error correction • Error detection and correction • Generate multiple alignments among overlapping reads • Identify instances where a base is overwhelmingly outvoted by bases aligned to it (taking into account the score quality) • Similarly correct occasional inserts and deletes (mostly due to sequencing errors)
ARACHNE: Evaluation of alignments • Evaluation of alignments • Assign a penalty score to each aligned pair of overlapping reads • Penalty scores are assigned to each discrepant base, based on the sequence quality score at the base and flanking bases on either side. • Discrepancies in high quality sequences are assigned high penalty, and discrepancies in low quality sequences are penalized less heavily. • The penalty scores for individual discrepancies are combined to yield an overall penalty score for the alignment. • Overlaps incurring too high a penalty are discarded • Likely chimeric reads are also detected and discarded • Reads that contain genomic sequence from two disparate locations are termed chimeric.
ARACHNE: paired pairs • Identification of paired pairs • Paired reads: reads which are known to be related with respect to orientation and distance. • Searches for instances of two plasmids of similar insert size with sequence overlap occurring at both ends. (together called paired pairs) • These instances are extended by building complexes of such pairs • *********** ********* • ********** ********** • ********* ******** • Collection of paired pairs are merged together into contigs.
ARACHNE: Contig assembly • When repeats are absent: correct assembly can be easily obtained by merging all the overlapping reads. • In presence of repeats, false overlaps may arise between reads derived from different copies of a repeat • ARACHNE identifies potential repeat boundaries and avoids assembling contigs across such boundaries • Potential repeat boundary: a read r can be extended by x and y, but x and y don’t overlap • Merge overlapping read pairs that do not cross a marked repeat boundary.
ARACHNE: repeat contigs and supercontigs • Detection of repeat contigs – identified 2 ways • Unusually high depth of coverage • Conflicting links to multiple, distinct, non-overlapping contigs, reflecting the multiple regions that flank the repeat in the genome. • aRb, cRd, eRf … will result in –aR-, -cR-, … • Creation of supercontigs • After marking repeat contigs the unmarked contigs (called unitigs) are assembled. • Use forward-reverse links from reads to order and orient unique contigs into supercontigs
ARACHNE: Filling gaps in supercontigs • Layout is a set of contigs each of which is an ordered list of contigs with interleaved gaps: corresponding to 2 kind of regions • Regions marked as repeat contigs (which were omitted in supercontig construction) • Regions for which there are insufficient number of shotgun reads to allow assembly • Fill gap using repeat contigs • For every pair of consecutive contigs with an interleaving gap in a supercontig S, the program tries to find a path of pairwise overlapping contigs that fill the gap. • Forward-reverse links from S guide the construction of the path by identifying contigs likely to fall in the gap.
Consensus derivation and postconsensus merger • The layout of overlapping reads is converted into consensus sequence with quality scores. • Done by converting pair-wise alignments of reads into multiple alignments, and deriving the consensus base by weighed voting.