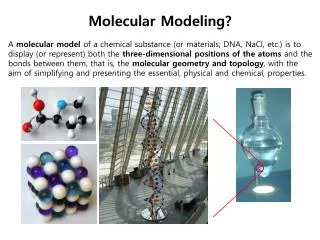
Molecular Modeling
Molecular Modeling. The compendium of methods for mimicking the behavior of molecules or molecular systems. Points for Consideration. Remember Molecular modeling forms a model of the real world Thus we are studying the model, not the world


Molecular Modeling
E N D
Presentation Transcript
Molecular Modeling The compendium of methods for mimicking the behavior of molecules or molecular systems
Points for Consideration • Remember • Molecular modeling forms a model of the real world • Thus we are studying the model, not the world • A model is valid as long as it reproduces the real world
? probe reactants products Why Use Molecular Modeling?(and not deal directly with the real world?) • Fast, accurate and relatively cheap way to: • Study molecular properties • Rationalize and interpret experimental results • Make predictions for yet unstudied systems • Study hypothetical systems • Design new molecules • And • Understand
Energy • Free energy (DG) • Enthalpy (DH) • Entropy (DS) • Steric energy (DG) • Molecular Spectroscopy • NMR (Nuclear Magnetic Resonance) • IR (Infra Red) • UV (Ultra Violate) • MW (Microwave) • 3D Structure • Distances • Angles • Torsions • Kinetics • Reaction Mechanisms • Rate constants • Electronic Properties • Molecular orbitals • Charge Distributions • Dipole Moments Some Molecular Properties
C7H5NO3S • A single 1D structure • A single 2D structure • Many 3D structures Molecular Structure: Saccharin
Property • Activity • Cell Permeability • Toxicity Structure • Descriptors • 1D: e.g., Molecular weight • 2D: e.g., # of rotatable bonds • 3D: e.g., Molecular volume Molecular Structure and Molecular Properties
Ball and spring description of molecules Better representation of equilibrium geometries than plastic models Able to compute relative strain energies Cheap to compute Lots of empirical parameters that have to be carefully tested and calibrated Limited to equilibrium geometries Does not take electronic interactions into account No information on properties or reactivity Cannot readily handle reactions involving the making and breaking of bonds Molecular mechanics
Polyatomic molecule: N-degrees of freedom N-dimensional potential energy surface Energy as a function of geometry http://www.chem.wayne.edu/~hbs/chm6440/PES.html
stretch bend torsion A Molecule is a Collection of Atoms Held Together by Forces • Intuitively forces act between bonded atoms • Forces act to return structural parameters to their equilibrium values
Stretch-bend Non-bonded And More Forces… • Forces also act between non-bonded atoms • Cross terms couple the different types of interactions
A Force Field is a Collection of Potential Energy Functions • A force field is defined by the functional forms of the energy functions and by the values of their parameters.
Molecular Mechanics Energy (Steric Energy) is Calculated from the Force Field • A molecular mechanics program will return an energy value for every conformation of the system. • Steric energy is the energy of the system relative to a reference point. This reference point depends on the bonded interactions and is both force field dependent and molecule dependent. • Thus, steric energy can only be used to compare the relative stabilities of different conformations of the same molecule and can not be used to compare the relative stabilities of different molecules. • Further, all conformational energies must be calculated with the same force field.
Steric Energy Steric Energy = Estretch +Ebend + Etorsion +EVdW +Eelectrostatic +Estretch-bend + Etorsion-stretch + … • Estretch Stretch energy (over all bonds) • Ebend Bending energy (over all angles) • Etorsion Torsional (dihedral) energy (over all dihedral angles) • EVdW Van Der Waals energy (over all atom pairs > 1,3) • Eelectrostatic Electrostatic energy (over all charged atom pairs >1,3) • Estretch-bend Stretch-bend energy • Etorsion-stretch Torsion-stretch energy • EVdW + Eelectrostatic are often referred to as non-bonded energies
energy coordinates Force Field and Potential Energy Surface • A force field defines for each molecule a unique PES. • Each point on the PES represents a molecular conformation characterized by its structure and energy. • Energy is a function of the coordinates. • Coordinates are function of the energy.
Moving on (Sampling) the PES • Each point on the PES is represents a molecular conformation characterized by its structure and energy. • By sampling the PES we can derive molecular properties. • Sampling energy minima only (energy minimization) will lead to molecular properties reflecting the enthalpy only. • Sampling the entire PES (molecular simulations) will lead to molecular properties reflecting the free energy. • In both cases, molecular properties will be derived from the PES.
Force Fields: General Features • Force field definition • Functional form (usually a compromise between accuracy and ease of calculation. • Parameters (transferability assumed). • Force fields are empirical • There is no “correct” form of a force field. • Force fields are evaluated based solely on their performance. • Force field are parameterized for specific properties • Structural properties • Energy • Spectra
In molecular mechanics atoms are given types - there are often several types for each element. • Atom types depend on: • Atomic number (e.g., C, N, O, H). • Hybridization (e.g., SP3, SP2, SP). • Environment (e.g., cyclopropane, cyclobutane). C O MM2 type 2 C C MM2 type 3 • “Transferability” is assumed - for example that a C=O bond will behave more or less the same in all molecules. Force Fields: Atom Types
A General Force Field Calculation • Input • Atom Types • Starting geometry • Connectivity • Energy minimization / geometry optimization • Calculate molecular properties at final geometry • Output • Molecular structure • Molecular energy • Dipole moments • etc. etc. etc.
Bonds • C-C x 2 • C-H x 8 • Angles • C-C-C x 1 • C-C-H x 10 • H-C-H x 7 • Torsions • H-C-C-H x 12 • H-C-C-C x 6 • Non-bonded • H-H x 21 • H-C x 6 A Simple Molecular Mechanics Force Field Calculation
Bond Stretching • Morse potential E r De = Depth of the potential energy minimum l0 = Equilibrium bond length (v(l)=0) w = Frequency of the bond vibration m = Reduced mass k = Stretch constant • Accurate. • Computationally inefficient (form, parameters). • Catastrophic bond elongation.
Harmonic potential (AMBER) E r Bond Stretching • Inaccurate. • Coincides with the Morse potential at the bottom of the well. • Computationally efficient.
Cubic (MM2) and quadratic (MM3) potentials harmonic cubic quadratic Bond Stretching
Bond Stretching Parameters (MM2) • Hard mode. • Bond types correlate with l0 and k values. • A 0.2Å deviation from l0 when k=300 leads to an energy increase of 12 kcal/mol.
AMBER: • MM2: • MM3: MM3 MM2 AMBER energy angle Angle Bending
Angle Bending Parameters (MM2) • Hard mode (softer than bond stretching)
Torsional (Dihedral) Terms • Reflect the existence of barriers to rotation around chemical bonds. • Used to set the relative energies of the rotational minima and maxima. • Together with the non-bonded terms are responsible for most of the structural and energetic changes (soft mode). • Usually parameterized last. • Soft mode.
General Functional Form • w: Torsional angle. • n (multiplicity): Number of minima in a 360º cycle. • Vn: Correlates with the barrier height. • g(phase factor): Determines where the torsion passes through its minimum value. Vn = 4, n = 2, g = 180 Vn = 2, n = 3, g = 60
Functional Form: AMBER • Preference to single terms. • Usage of general torsional parameters (i.e., torsional potential solely depends on the two central atoms of the torsion). • C-C-C-C = O-C-C-C = O-C-C-N = …
# Type V1 V2 V3 1 C-C-C–C 0.200 0.270 0.093 4 C-C-C–H 0.000 0.000 0.267 4 H-C-C–H 0.000 0.000 0.237 Example: Butane • The barrier to rotation around the C-C bond in butane is ~20kJ/mol. • All 9 torsional interactions around the central C-C bond should be considered for an appropriate reproduction of the torsional barrier.
O O Out-of-Plane Bending • Allows for non-planarity when required (e.g., cyclobutanone). • Prevents inversion about chiral centers (e.g., for united atoms). • Induces planarity when required. correct • United Atoms • Absorb non-polar hydrogen atoms into respective carbons. • Greatly reduces computational cost.
j l i k i j l k Out-of-Plane Bending • Wilson Angle Between ijk plane and i-l bond • Pyramidal Distance Between atom i and jkl plane • Improper torsion 1-2-3-4 torsion
Cross Terms: Stretch-Bend • Coupling between internal coordinates. • Important for reproducing structures of unusual (e.g., highly strained ) systems and of vibrational spectra. Stretch-Bend: As a bond angle decreases, the adjacent bonds stretch to reduce the interaction between the 1,3 atoms.
Cross Terms: Stretch-Torsion Stretch-Torsion: For an A-B-C-D torsion, the central B-C bond elongates in response to eclipsing of the A-B and C-D terminal bonds.
Non-Bonded Interactions • Operate within molecules and between molecules. • Through space interactions. • Modeled as a function of an inverse power of the distance. • Soft mode. • Divided into: • Electrostatic interactions. • VdW interactions.
Electrostatic Interactions • qi, qj are point charges. • Charge-charge interactions are long ranged (decay as r-1). • When qi, qj are centered on the nuclei they are referred to as partial atomic charges. • Fit to known electric moments (e.g., dipole, quadrupole etc.) • Fit to thermodynamic properties. • Ab initio Calculations • Electrostatic potential
Rapid Methods for Calculating Atomic Charges • Partial Equalization of Orbital Electronegativity (Gasteiger and Marsili) • http://www2.chemie.uni-erlangen.de/software/petra/manual/manual.pdf • Electronegativity (Pauling): • “The power of an atom to attract an electron to itself” • Orbital electronegativity depends upon: • Valence state (e.g., sp > sp3). • Occupancy (e.g., empty > single > double). • Charges in other orbitals. • Electrons flow from the less electronegative atoms to the more electronegative atoms thereby equalizing the electronegativities.
Partial Equalization of Orbital Electronegativity • For the dependency of orbital electronegativity on charge assume: • Theoretically correct for orbitals but formally applied to atoms. • Values of a, b and c were obtained for common elements in their usual valence states (e.g., for atom types). • Apply an interactive process: • Assign each atom its formal charge. • Calculate atomic electronegativities based on above assumption. • Calculate the electron charge transferred from atom A to the more electronegative atom B bonded to it by: Dumping factor Cation Electronegativity of the less electronegative atom
Solvent Dielectric Models • e0 = Dielectric constant of vacuum (1). • For a given set of charges and distance, e0 determines the strength of the electrostatic interactions. • Solvent effect dampen the electrostatic interactions and so can be modeled by varying e0: • eeff = e0er • er(protein interior) = 2-4 • er(water) = 80
Dielectric constant increases as a function of distance • Smooth increase in eeff form 1 to er as the distance increases eeff distance Solvent Dielectric Models: Distance Dependence eeff varies from 1 at zero separation to the bulk permitivity of the solvent at large separation. As the separation between the interacting atoms increases, more solvent can “penetrate” between them.
Van Der Waals (VdW) Interactions • Electrostatic interactions can’t account for all non-bonded interactions within a system (e.g., rare gases). • VdW interactions: • Attractive (dispersive) contribution (London forces) • Instantaneous dipoles due to electron cloud fluctuations. • Decays as r6. • Repulsive contribution • Nuclei repulsion. • At short distances (r < 1) rises as 1/r. • At large distances decays as exp(-2r/a0); a0 the Bohr radius.
Van Der Waals (VdW) Interactions • The observed VdW potential results from a balance between the attractive and repulsive forces.
Lennard-Jones Potential • Rapid to calculate • Attractive part theoretically sound • Repulsive part easy to calculate but too steep s energy e • Alternative forms rm separation Modeling VdW Interactions
Don w Acc C O q N H Hydrogen Bonding • H-bond geometry independent: • r is the distance between the H-bond donor and H-bond acceptor. • H-bond geometry dependent (co-linearity with lp preferred):
Force Field Parameterization • Choosing values for the parameters in the potential function equations to best reproduce experimental data. • Parameterization techniques • Trial and error • Least square methods • Types of parameters • Stretch: natural bond length (l0) and force constants (k). • Bend: natural bond angles (q0) and force constants (k). • Torsions: Vi’s. • VdW: (e, VdW radii). • Electrostatic: Partial atomic charges. • Cross-terms: Cross term parameters.
Required Actual Stretch Bend Torsion 1521 58,319 2,313,441 164 357 508 Parameters - Quantity • For N atom types require: • N non-bonded parameters • N*N stretch parameters • N*N*N bend parameters • N*N*N*N torsion parameters • Example • MacroModel MM2*: 39 atom types
Parameters - Source • A force field parameterized according to data from one source (e.g., experimental gas phase, experimental solid phase, ab initio) will fit data from other sources only qualitatively. • Experiment: geometries and non-bonded parameters • X-Ray crystallography • Electron diffraction • Microwave spectroscopy • Lattice energies • Advantages • Real • Disadvantages • Hard to obtain • Non-uniform • Limited availability