Computational problems involving Single Nucleotide Polymorphisms
Computational problems involving Single Nucleotide Polymorphisms. Pritam Chanda. Agenda. Biological background SNP representation Tag SNP selection Haplotype analysis SNP-disease association study Discussion. Central Dogma. A cell and its chromosomes. DNA structure.


Computational problems involving Single Nucleotide Polymorphisms
E N D
Presentation Transcript
Computational problems involving Single Nucleotide Polymorphisms Pritam Chanda
Agenda • Biological background • SNP representation • Tag SNP selection • Haplotype analysis • SNP-disease association study • Discussion
DNA structure Base pairs : A-T, G-C A T A T A T G C A G C A 3’ Template strand 5’ 3’ 5’ T A T A T A C G T C G T Thus, each chromosome can be thought of as a sequence of A, T, G, C’s Anti-parallel chain
Genomic Variation and SNPs • Human Genome 3 109 base pairs. • Main form of variation between individual genomes: single nucleotide polymorphisms (SNPs) • Single base changes in the genome sequence that occurs in a significant proportion (more than 1 percent) of the population • Most SNPs are bi-allelic (two variations) • Total #SNPs 1 107 • Difference between any two individuals 3 106 SNPs ( 0.1% of entire genome) Sequences on a pair of homologous chromosomes
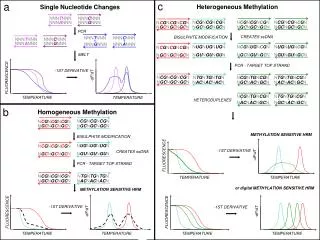
Why important ? • A SNP (pronounced as ‘snip’) can alter the amino acid sequence of the protein produced. • Not always • A protein consists of sequence of amino acids. • There are total 20 amino acids • Genetic code produces amino acids by reading groups of 3 nucleotides at a time • 43 combinations = 64 different combinations of A,T,G,C. • Thus not all combinations of 3 nucleotides produce different amino acids • Redundancy in genetic code. • A SNP in which both alleles lead to the same protein sequence is termed synonymous • If different proteins are produced they are non-synonymous.
Why important ? • SNPs that are not in protein coding regions may still have consequences for gene splicing, transcription factor binding, or the sequence of non-coding RNA. • SNPs in humans can affect how humans develop diseases, respond to pathogens, chemicals, drugs, etc. • SNPs are inherited and do not change much from generation to generation in an individual with time, • SNPs are of great value to biomedical research and in developing diagnostic and pharmaceutical products.
A G A T A G T A A T A G A T C G T A A T A G A T A G T A A T A G A T A G T A A T A G A T A G T A A T A G A T C G T A A T A G A T 0 G T A A T A G A T 1 G T A A T A G A T 0 G T A A T A G A T 0 G T A A T A G A T 0 G T A A T A G A T 1 G T A A T Sample 1 Sample 1 Sample 2 Sample 2 Sample 3 Sample 3 Bioinformatics representation Sequences on a pair of homologous chromosomes • Assumption: a snp is bi-allelic. • Major allele • most frequent allele • Minor allele • The other one • Example • Given DNA sequence • Major allele (A) - 67% • Minor allele (C) - 33% • Encoding • Major allele : 0 • Minor allele : 1
snps Other nucleotides A G A T A G T A A T A C A T GG T A A A Haplotypes and Genotypes • Diploid organisms: cells have two homologous set of chromosomes. • Haplotype: description of SNP alleles on a single chromosome • 0/1 vector, e.g., 00110101 (here, 0 is for major, 1 is for minor allele). • Genotype: combined description of SNP alleles on pairs of homologous chromosomes • 0/1/2 vector, e.g., 01122110 (0=0+0, 1=1+1, 2=0+1 or 1+0) • Each genotype with k 2’s (heterozygotes) can be explained by 2k-1 pairs of haplotypes Haplotype Genotype 0 1 0 1 1 1 00 1 0 2 2 0 1 2 Major allele Minor allele Heterozygous Homozygous
SNP databases • HapMap project (www.hapmap.org) • The aim of the project is to record the significant SNPs. • Started in October 2002. • Phase 1 data have been published and analysis of Phase 2 data is underway as of October 2006. • dbSNP • A database of SNPs and short deletion and insertion polymorphisms at NCBI. • CGAP • Genetic variation in genes important in cancer (At the National Cancer Institute) • EnsEMBL • Joint project between EMBL-EBI and the Sanger Centre to develop a system which produces and maintains automatic annotation on eukaryotic genomes. • The SNP Consortium • Information about up to 300000 SNPs. • Many more…
Linkage Disequilibrium (LD) Haplotype Frequency • LD measures the correlation between two SNPs. • Some combinations of alleles or genetic markers occur more or less frequently in a population than would be expected from a random formation of haplotypes from alleles based on their frequencies. • Non-random associations between genes at different loci are measured by the degree of linkage disequilibrium (D). • Consider two loci case (i.e. two SNPs) • SNP1 has alleles A, a • SNP2 has alleles B, b • When the two loci are independent, expected freq of haplotype AB is pAB = pApB • LD measure: D = pAB - pApB Allele Frequency
LD measures • D D = pAB – pApB, pAB = pApB + D pAb = pA – pAB = pA – pApB – D = pA(1-pB) – D = pApb – D • D’ = D/Dmax • r2 = D/(pApapBpb)
Types of Diseases Monogenic & Complex Diseases • Monogenic diseases – rarer (<0.1%) • Mutated gene is entirely responsible for the disease • Easy to locate diseased gene using LD based association studies. • Complex diseases (more common) • Interaction of multiple genes in a complicate fashion • One mutation does not cause disease • Hard to analyze – a single SNP may show weak association • A specific combination may show strong association, but what combination ? • Multiple independent causes • There are different causes and each of these causes can be result of interaction of several genes • Each cause explains a certain percentage of cases
Tag SNP • SNPs are inherited from one generation to another in blocks. • Each block contains a few common haplotypes and the SNPs in the block are in LD. • Because of LD, each block contains a minimal informative set of SNPs that can represent the rest of the SNPs with high accuracy and also can identify all the haplotypes of the block. • Tag SNPs. • Study of genetic factors for complex diseases • Several genes contribute together to the disease. • Need to study a relatively large number of SNPs. • Also need a bigger sample size of individuals.
Tag SNP problem definition • Genotyping a large number of SNPs is cost-prohibitive. • Essential to choose a set of SNPs to be genotyped such that this set predicts the rest of the SNPs (not typed) with high accuracy. • This set of SNPs is called the tag SNPs. • Tag SNP selection deals with finding a set of tag SNPs of minimum size that would have very good prediction ability for the rest of the SNPs.
LD based tag SNP selection • Greedy algorithm to identify subsets of tagSNPs for genotyping • Start with all SNPs above a MAF threshold and calculate pair-wise LD. • Select the SNP that exceeds a LD threshold with the maximum number of other sites. • This maximally informative SNP and all associated SNP are grouped as a bin of associated sites. • All pairwise LD within bin are re-evaluated, and any SNP exceeding threshold LD with all other sites in the bin is specified as a tagSNP for the bin. • Repeat the bining process analyzing all as-yet-unbinned SNPs at each round, until all sites exceeding the MAF threshold are binned. • If an SNP does not exceed the LD threshold with any other SNP in the region, it is placed in a singleton bin.
Tag SNP using feature selection • Given N x M matrix • N haploid sequences • M snps • Each snp is a feature. • Select the minimum set of features to classify all haplotypes accurately. • r2 = (pABpab – pAbpaB)/(pABpAbpaBpab) • FSFS selects the most informative set of SNPs by first grouping them into homogenous subsets and then choosing a representative SNP from each group. • Designed only for haplotypes Phuong T. M., Lin Z., Altman R. B. Choosing SNPs Using Feature Selection. Proc IEEE Comput Syst Bioinform Conf. 2005; 301-9.
Feature selection algorithm • Let, set of all SNPs : S = {F1; F2; ...;FN}. • D(Fi; Fj) represents the dissimilarity between the two SNPs (Fi and Fj ) and is calculated using r2. • R represent the final set of SNPs chosen as the tag SNPs. • FSFS takes as input S and K (# of nearest neighbors of a SNP to consider), • During each iteration, FSFS calculates the distance D(i,k) between each SNP F(i) in R and its kth nearest neighboring SNP. • The algorithm then finds SNP F0 for which D(0,k) is minimum, retains this SNP in R and removes its K nearest SNPs from R. • Thus the algorithm always discards SNPs from the most compact cluster causing the minimum information loss. • FSFS gradually decreases K and re-computes D(0,k) until D(0,k) is less than or equal to a threshold. Phuong T. M., Lin Z., Altman R. B. Choosing SNPs Using Feature Selection. Proc IEEE Comput Syst Bioinform Conf. 2005; 301-9.
k snps 0 1 0 1… 1 1 0 0 1… 0 …………… 1 1 0 1… 1 1 0 1 .. M samples 1 1 … .. 0 s A Regression based method • Uses Multivariate Linear Regression (MLR) • SNP value prediction • (n+1)x(k+1) matrix M corresponding to n sample individuals and the individual x and k tag SNPs (assume already known for prediction purpose) and a single non-tag SNP s (whose value the tag SNPs will predict). • All SNP values in M are known except the value of s in x. • In case of haplotypes, there are only two possible resolutions of s, s0 (for SNP value 0) and s1 (for SNP value 1). • For genotypes, there are 3 possible resolutions s0 (SNP value 0), s1 (SNP value 1), and s2 (SNP value 2). • The SNP prediction method should predict correct resolution of s. Jingwu H. and Zelikovsky A. Tag SNP Selection Based on Multivariate Linear Regression. Proc. of Intl Conf on Computational Science (ICCS 2006), May 2006, LNCS 3992, pp. 750-757.
MLR • The set of tag SNPs T are vectors in the (n+1)-dimensional Euclidean space. • Get the projections of the vectors s0, s1 and s2 onto the span of the set of tag SNPs. • The most probable resolution of s should be closest to the span of T. • A Greedy Algorithm • Start with selecting the best tag t0 that alone predicts all other tags with minimum prediction error, • In each iteration, continue to add tags to the set T such that T best predicts the remaining tags. Jingwu H. and Zelikovsky A. Tag SNP Selection Based on Multivariate Linear Regression. Proc. of Intl Conf on Computational Science (ICCS 2006), May 2006, LNCS 3992, pp. 750-757.
Other methods • Entropy based methods • Support vector machines • Bayesian methods • Principal Component analysis Haplotype tagging using support vector machines. Granular Computing, 2006 IEEE International Conference on. Jingwu He; Jun Zhang; Altun, G.; Zelikovsky, A.; Yanqing Zhang Page(s): 758- 761 Haplotype Block Partitioning and Tag SNP Selection Using Genotype Data and Their Applications to Association Studies - Kui Zhang, Zhaohui S. Qin, Jun S. Liu, Ting Chen, Michael S. Waterman and Fengzhu Sun Genome Research 14:908-916, 2004 Lin Z., Altman R. B. Finding haplotype tagging SNPs by use of principal components analysis. Am J Hum Genet. 2004 Nov;75(5):850-61. Hampe J., Schreiber S., Krawczak M. Entropy-based SNP selection for genetic association studies. (2003) Hum Genet 114:36-43.
0 1 1 1 0 0 1 1 0 1 1 0 1 0 0 1 0 0 Two haplotypes per individual Merge the haplotypes 2 1 2 1 0 0 1 2 0 Genotype for the individual Haplotype Estimation • Each individual has two “copies” of each chromosome. • At each site, each chromosome has one of two alleles (states) denoted by 0 and 1 (0 major allele, 1 = minor allele) • HapMap Project • NIH lead project ($100M) to find common haplotypes in the Human population. • Haplotyping individuals is expensive.
Haplotyping issues • Biological Problem: For disease association studies, haplotype data is more valuable than genotype data, but haplotype data is hard to collect. Genotype data is easy to collect. • Computational Problem: Given a set of n genotypes, determine the original set of n haplotype pairs that generated the n genotypes. 2 0 1 2 Genotype 0 0 1 0 1 0 1 1 1 0 1 0 0 0 1 1 Possible valid Haplotypes Each genotype with k 2’s (heterozygotes) can be explained by 2k haplotypes
Need for haplotype inference • Why do we want to determine haplotypes for individuals at tightly linked SNP loci? • Haplotypes are more powerful discriminators between cases and controls in disease association studies. • With haplotypes we can conduct evolutionary studies. • Use of haplotypes in disease association studies reduces the number of tests to be carried out, and hence the penalty for multiple testing. • Two aspects of the problem • Estimate the frequencies of all possible haplotypes in the population. • Infer the haplotypes of all individuals in the given sample.
Clark’s method • Haplotype inference by A. Clark in 1990. • With a reasonable sample size, we expect to have some individuals homozygous at every locus, e.g. 1—0—1, or heterozygous at just one locus, e.g. 1—0—2. • For the first case, unambiguously identify haplotype (1—0—1), • From the second case, two (1—0—2 and 1—0—1) haplotypes are present in the population. • The algorithm begins by finding all homozygotes and single SNP heterozygotes and tallying the resulting known haplotypes. • For each known haplotype, check if the known haplotype can be made from some combination of ambiguous sites from an unresolved case. • 1—0—1 known . So resolve 2—0—2 as (1—0—1) + (0—0—0). • This chain of inferences is continued until either all haplotypes have been recovered, or until no more new haplotypes can be found in this way.
Hardy Weinberg Equilibrium • Consider a SNP with two alleles A,a • 3 possible genotypes A/A, A/a and a/a. • pA, pa are the individual allele frequencies. • HWE assumes that a child inherits the two alleles independently from his parents. • A population in which A/A occurs with probability p2A, A/a with 2pApa and a/a with p2b is said to be in HWE. • Under a certain set of assumptions like infinite population size, random mating etc, the genotype frequencies stabilize.
Maximum Likelihood Estimation • Given a SNP with alleles M, m. • Possible genotypes are M/M, M/m, m/m. • What is the probability of seeing a M/M’s, b M/m’s and c m/m’s ? • According to HWE, probability that any one particular individual selected is MM, Mm or mm is pM2, 2pMpm, pm2. • Taking log, differentiating and setting to 0 gives the maximum likelihood estimates • pM = (2a+b)/2N, pm = (2c+b)/2N
Data (D) Available Data Missing Data θ = Parameters to calculate the missing data Expectation Maximization (EM) • E-step • The missing data is calculated using θ. This along with the available data forms the complete data (D). • M-step • θ’ = Recalculate the maximum likelihood estimates of θ from D. Repeat E-step with θ= θ’.
Using EM • Consider a 2-loci case • Bi-allelic loci • So possible haplotypes • AB, Ab, aB, ab. • We are given observed counts of each possible genotype • 9 possible genotypes • AABB, AABb, AAbb, AaBB, … • Observe that only genotype AaBb can have more than 2 different haplotypes x 1-x x = fraction of genotype AaBb that are
Using EM Parameters = pAB, pAb, paB, pab (haplotype frequencies) • Calculate pAB etc. from given genotype frequencies. • The allele frequencies are • pA = (30+73/2)/129 = 0.5155 • pa = (26+73/2)/129 = 0.4845 • pB = (23+78/2)/129 = 0.4806 • pb=(28+78/2)/129 = 0.5194 • The haplotype frequencies are • pAB=[2(10)+15+10+50x]/[129(2)] • pAb=[15+2(5)+50(1-x)+13]/[129(2)] • paB=[50x+3+13+28(2)]/[129(2)] • pab=[50(1-x)+13+13+10(2)]/[129(2)] • The problem is to estimate the 4 haplotype frequencies despite not knowing the value of x (our missing data).
E-step • E-step : obtain some initial values for the haplotype frequencies • Assume we have simply each genotype frequency as product of the respective allele frequencies. • p0AB = (0.5155)(0.4806) • p0Ab = (0.5155)(0.5194) • p0aB = (0.4845)(0.4806) • p0ab = (0.4845)(0.5194) • The ‘expected’ value of x given these haplotype frequencies, is
M-step • M-step : maximize the parameters (haplotype frequencies) using x0 calculated at the E-step. • Substitute x0 into the haplotype frequencies. • p1AB = [2(10)+15+10+50x]/[129(2)] = 0.27131 • p1Ab = [15+2(5)+50(1-x)+13]/[129(2)] = 0.24418 • p1aB = [50x+3+13+28(2)]/[129(2)] = 0.20930 • p1ab = [50(1-x)+13+13+10(2)]/[129(2)] = 0.27519 • Repeat E-step and M-step until the haplotype frequencies do not change much.
Other methods • Bayesian methods • Combinatorial methods • Dynamic programming Haplotype Block Partitioning and Tag SNP Selection Using Genotype Data and Their Applications to Association Studies Kui Zhang, Zhaohui S. Qin, Jun S. Liu, Ting Chen, Michael S. Waterman and Fengzhu Sun Genome Research 14:908-916, 2004 V. Bafna, D. Gusfield, G. Lancia, and S. Yooseph. Haplotyping asperfect phylogeny: A direct approach. Technical report, UC Davis,Department of Computer Science, 2002. Bayesian Haplotype Inference via the Dirichlet Process, Xing et. al, in Proceedings of the Second RECOMB Satellite Workshop on Computational Methods for SNP and Haplotypes, pp. 99-112; An Entropy-Based Statistic for Genomewide Association Studies Jinying Zhao,Eric Boerwinkle,and Momiao Xiong Am J Hum Genet. 2005 July; 77(1): 27–40.
Support Vector Machines • Given training set of instance-label pairs (xi,yi), i = 1,... , L where xiεRn and yε {1,−1}L, the (SVM) seeks solution to the following optimization problem: • Training vectors xi are mapped into a higher dimensional space by the function Φ. • SVM finds a linear separating hyper-plane with the maximalmargin in this higher dimensional space. • C > 0 is the penalty parameter of the errorterm.
Support Vector Machines • SVM machine for binary classification. The margin to be maximized is w that separates the hyper-plane (shown with dotted line) from the two classes of data.
Multiple Myeloma (a type of cancer) is studied. • The data set consists of genotypes from 3000 SNPs for 80 patients selected so that they are evenly spaced at about 1Mb apart to give a good overall coverage of the human genome. • Each heterozygous SNP data is coded as 0, one homozygous is arbitrarily coded as +1 and the other as -1. • Entropy based feature selection • Select the most informative top 10% SNPs from the set of 3000 SNPs. • The entropy of a data set is given by - p log2(p) - (1 - p) log2(1 - p) where p is the fraction of examples that belong to class predisposed. • The information gain of the split is given by the entropy of the original data set minus the weighted sum of entropies of the two data sets resulting from the split, where these entropies are weighted by the fraction of data points in each set. • The SNP features are ranked by information gain, and the top-scoring 0% of the features are selected. • Classification of the diseased and control cases using a leave-one-out cross validation approach yields an overall classification accuracy of 71% which is significantly better than chance (50%). Waddell M., Page D., Zhan F., Barlogie B. and John Shaughnessy Jr. J. Predicting Cancer Susceptibility from Single-Nucleotide Polymorphism Data: A Case Study in Multiple Myeloma, Proceedings of BIOKDD '05, Chicago, Illinois, August 2005, Aug 2005.
Disease Status Genotypes 1 2 3 4 5 6 7 8 9 10 0 1 0 1 2 0 1 0 2 0 2 0 1 1 0 2 1 0 1 2 1 2 0 0 1 2 2 2 1 0 1 1 0 1 2 0 2 0 2 0 2 0 1 2 0 0 1 0 1 2 2 0 0 0 2 0 2 1 0 0 2 1 0 1 1 0 0 0 2 1 1 1 1 2 2 2 2 Healthy genotypes (Control) Diseased Genotypes (Case) A Combinatorial approach Case/Control study Given :A population of n genotypes each containing values of m SNPs and disease status. 0: homozygous major allele, 1: homozygous minor allele, 2 : heterozygous allele Disease association analysis searches for risk (resistance) factor with frequency among case (control) individuals considerably higher than among control (case) individuals.
1 2 3 4 5 6 7 8 9 status 0 1 1 0 1 2 1 0 2 case 0 1 1 1 0 2 0 0 1 case 0 0 1 0 0 0 0 2 1 case 0 1 1 1 1 2 0 0 1 case 0 0 1 0 1 2 1 0 2 control 0 1 0 0 1 1 0 0 2 control 0 1 1 0 1 2 0 0 2 control Multi-SNP Combination (MSC)[1,2] Multi-SNP extension • Snp(C) : subset of given SNPs. • MSC(C) : a specific value of Snp(C). • Cluster(C) : subset of individuals that coincides with {Snp(C), MSC(C)} in the given genotype data. 1234567 C = (1,2,4,5,7) D(C) = (1,2,4) H(C) = (5,7) Snp(C) = (3,6) x x 1 x x 2 x x x MSC(C) present in 4 cases : 1 control How significant is this cluster ? [1] Combinatorial Search Methods for Multi-SNP Disease Association. Brinza et. al., 2006. [2] Combinatorial Methods for Disease Association Search and Susceptibility Prediction. Brinza et. al., 2006.
P-value of MSC[1,2] • Measured P-value • Probability that diseased/healthy distribution among exposed to risk factor happened by chance • Compute by binomial distribution • Searching for risk factors among many SNPs requires multiple testing adjustment of the p-value [1] Combinatorial Search Methods for Multi-SNP Disease Association. Brinza et. al., 2006. [2] Combinatorial Methods for Disease Association Search and Susceptibility Prediction. Brinza et. al., 2006.
Disease Association problem formulation Given:Each containing values of m SNPs and disease status Case/control study data consisting of n genotypes Find:All Risk/Resistance factors (MSCs) with p-value below 0.05
i j j i 0 1 1 0 1 2 1 0 2 case 0 1 1 0 1 2 1 0 2 case 2 0 1 1 0 2 0 0 2 case 2 0 1 1 0 2 0 0 2 case 0 0 1 0 0 0 0 2 1 case 0 0 1 0 0 0 0 2 1 case 0 1 1 0 1 2 0 0 2 control 0 1 1 0 1 2 0 0 2 control Case-closure 0 1 1 0 1 2 0 1 2 control 0 1 1 0 1 2 0 1 2 control x x 1 x x 2 x x x MSC MSC’ x x 1 x x 2 x 0 x Present in 2 cases : 2 controls Present in 2 cases : 1 controls Searching Approaches Exhaustive search (ES)[1,2] • Computationally infeasible, exponential number of combinations • Searching for 3-SNP MSC on the sample with n genotypes and m SNPs requires O(n3m) • Case-closureof a MSC C is an MSC C’, with maximum number of SNPs with fixed values, which consists of the same set of cases and minimum number of controls. • Efficient way for finding case-closure: Extend MSC with those SNPs that have common values in all cases. Cluster C:subset of genotypes which share the same MSC [1] Combinatorial Search Methods for Multi-SNP Disease Association. Brinza et. al., 2006. [2] Combinatorial Methods for Disease Association Search and Susceptibility Prediction. Brinza et. al., 2006.
Combinatorial Search Combinatorial search (CS)[1,2] • Combinatorial Search Method (CS) • Searches only among case-closed MSCs • Avoids checking of clusters with small number of cases • Finds significant MSCs faster than ES • Still too slow for large data • Further speedup by reducing number of SNPs • Indexing:compress S by extracting most informative SNPs • Tag SNP Selection • Apply ES/CS on selected tag snps [1] Combinatorial Search Methods for Multi-SNP Disease Association. Brinza et. al., 2006. [2] Combinatorial Methods for Disease Association Search and Susceptibility Prediction. Brinza et. al., 2006.
Discussion • Neural networks, hidden markov models, interaction information, linkage analysis etc. • In general machine learning methods tend to do better than purely combinatorial methods and also are applicable to bigger data sets with hundreds of SNPs. • Scalablity • Identifying SNPs in disease association study is more difficult, largely depends on the population under study and often faces the problem of replication.