Mass Spectrometry-based Proteomics
Mass Spectrometry-based Proteomics. Xuehua Shen (Adapted from slides with textbook). Outline. Motivation of proteomics Mass spectrometry-based proteomics Instrumentation of mass spectrometry De novo sequencing algorithm Database search Algorithms of real software (e.g., sequence tags).


Mass Spectrometry-based Proteomics
E N D
Presentation Transcript
Mass Spectrometry-based Proteomics Xuehua Shen (Adapted from slides with textbook)
Outline • Motivation of proteomics • Mass spectrometry-based proteomics • Instrumentation of mass spectrometry • De novo sequencing algorithm • Database search • Algorithms of real software (e.g., sequence tags)
mRNA DNA Protein Alternative splicing Post-translational Modification SNP ~30,000 human genes >100,000 RNA messages >1,000,000 distinct protein forms Motivation • Proteins are working units of the cells • The number of found genes is much less than the number of expressed proteins • Directly related with cell processes and diseases
Tools for Proteomics • Edman degradation reaction • NMR (Nuclear Magnetic Resonance) • X-ray crystallography • Protein array • Mass Spectrometry
Mass Spectrometry-based Proteomics • Primary sequence (sequencing, identification) • Post-translational modification (PTM) (characterization) • Quantitative proteomics (quantification) • Protein-protein interaction
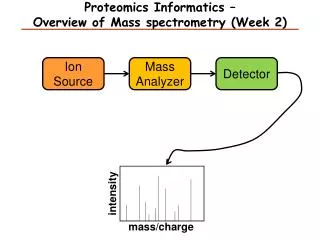
Components of Mass Spectrometer • Ion source (ESI and MALDI) • Mass analyzer (ion traps, TOF, Quadrupole, FT, etc.) • Mass-to-charge ratio (m/z) • Ion detector
Peptide and Intact Protein • Peptide: a fragment of protein • Some enzymes, e.g. trypsin, break protein into peptides. • Some technology put intact protein into the mass spectrometer
Peptide Fragmentation Collision Induced Dissociation H+ H...-HN-CH-CO . . .NH-CH-CO-NH-CH-CO-…OH Ri-1 Ri Ri+1 N-Terminus C-Terminus • Peptides tend to fragment along the backbone. • Fragments can also loose neutral chemical groups like NH3 and H2O.
N- and C-terminal Peptides P A G N F A P G N F A N P G F C-terminal peptides N-terminal peptides A N F P G P A N F G
Terminal peptides and ion types P G N F Peptide H2O Mass (D) 57 + 97 + 147 + 114 = 415 P G N F Peptide without H2O Mass (D) 57 + 97 + 147 + 114 – 18 = 397
N- and C-terminal Peptides 486 P A G N F A 71 P G N F 415 301 A N P G F 185 C-terminal peptides N-terminal peptides A N F P G 332 154 P A N F G 429 57
N- and C-terminal Peptides 486 71 415 301 185 C-terminal peptides N-terminal peptides 332 154 429 57
N- and C-terminal Peptides 486 71 415 301 185 332 154 429 57
N- and C-terminal Peptides 486 71 415 Problem: Reconstruct peptide from the set of masses of fragment 301 185 332 154 429 57
G V D L K L 57 Da = ‘G’ K D V G 99 Da = ‘V’ H2O D Mass Spectra • The peaks in the mass spectrum: • Prefix • Fragments with neutral losses (-H2O, -NH3) • Noise and missing peaks. mass 0 and Suffix Fragments.
G V D L K • Peptide Identification: Intensity MS/MS mass 0 mass 0 Protein Identification with MS/MS
Protein Identification by Tandem Mass Spectrometry S e q u e n c e MS/MS instrument • De Novo interpretation • Sherenga • Database search • Sequest
W R V A L Database ofknown peptidesMDERHILNM, KLQWVCSDL, PTYWASDL, ENQIKRSACVM, TLACHGGEM, NGALPQWRT, HLLERTKMNVV, GGPASSDA, GGLITGMQSD, MQPLMNWE, ALKIIMNVRT, AVGELTK, HEWAILF, GHNLWAMNAC, GVFGSVLRA, EKLNKAATYIN.. Database ofknown peptidesMDERHILNM, KLQWVCSDL, PTYWASDL, ENQIKRSACVM, TLACHGGEM, NGALPQWRT, HLLERTKMNVV, GGPASSDA, GGLITGMQSD, MQPLMNWE, ALKIIMNVRT, AVGELTK, HEWAILF, GHNLWAMNAC, GVFGSVLRA, EKLNKAATYIN.. T G E P L K C W D T W R V A L T G E P L K C W D T De Novo vs. Database Search Database Search De Novo Mass, Score AVGELTK
Current Status • It is still a open problem of protein sequencing no matter whether using de novo sequencing or database search methods • Following algorithms only deal with simplified (or ideal) spectrums • Some algorithms combine de novo sequencing and database search
Pros and Cons of de novo Sequencing • Advantage: • Gets the sequences that are not necessarily in the database. • An additional similarity search step using these sequences may identify the related proteins in the database. • Disadvantage: • Requires higher quality data. • Often contains errors.
Outline • Motivation of proteomics • Mass spectrometry-based proteomics • Instrumentation of mass spectrometry • De novo sequencing • Database search • Algorithms of real software (e.g., sequence tags)
De novo Peptide Sequencing Sequence
Peptide Sequencing Problem Goal: Find a peptide with maximal match between an experimental and theoretical spectrum. Input: • S: experimental spectrum • Δ: set of possible ion types • m: parent mass Output: • P: peptide with mass m, whose theoretical spectrum matches the experimental S spectrum the best
Procedure of De Novo Sequencing • Build spectrum graph • How to create vertices (from masses) • How to create edges (from mass differences) • Find best path or rank paths of spectrum graph • How to find candidate paths • How to score paths
From Sequence to Spectrum S E Q U E N C E b Mass/Charge (M/Z)
From Sequence to Spectrum(cont.) a S E Q U E N C E Mass/Charge (M/Z)
From Sequence to Spectrum (cont.) a is an ion type shift in b S E Q U E N C E Mass/Charge (M/Z)
From Sequence to Spectrum (cont.) y E C N E U Q E S Mass/Charge (M/Z)
From Sequence to Spectrum (cont.) Intensity Mass/Charge (M/Z)
From Sequence to Spectrum (cont.) Intensity Mass/Charge (M/Z)
From Sequence to Spectrum (cont.) noise Mass/Charge (M/Z)
MS/MS Spectrum Intensity Mass/Charge (M/z)
Some Mass Differences between Peaks Correspond to Amino Acids u q e e q s u e n n c e e e q c s n e u s e c e
Now decoding from spectrum to sequence…? Build spectrum graph
b2-H2O b3- NH3 a2 b2 a3 b3 HO NH3+ | | R1 O R2 O R3 O R4 | || | || | || | H -- N --- C --- C --- N --- C --- C --- N --- C --- C --- N --- C -- COOH | | | | | | | H H H H H H H y3 y2 y1 y2 - NH3 y3 -H2O Peptide Fragmentation • Different ion types (b, y, b-NH3, b-H2O) • Fragment at one site (internal ions)
Example of Ion Type • Δ={δ1, δ2,…, δk} • Ion types {b, b-NH3, b-H2O} correspond to Δ={0, 17, 18} *Note: In reality the δ value of ion type b is -1 but we will “hide” it for the sake of simplicity
Why Peptide Sequencing hard • Two ladders of overlapping masses, could not tell whether it is b ion or y ion • Incomplete fragmentation • Chemical noise • Mass accuracy of the instrument is not good enough (Q=K, G+V=156.090, R=156.101) • Q: Is sequencing shorter or longer peptide harder?
Vertices of Spectrum Graph • Vertices are generated by reverse shifts corresponding to ion typesΔ={δ1, δ2,…, δk} • Every mass s in an MS/MS spectrum generates k vertices V(s) = {s+δ1, s+δ2, …, s+δk} corresponding to potential N-terminal peptides • Vertices of the spectrum graph: {initial vertex}V(s1) V(s2) ... V(sm) {terminal vertex}
Reverse Shifts Shift in H2O Shift in H2O+NH3
Edges of Spectrum Graph • Two vertices with mass difference corresponding to an amino acid A: • Connect with an edge labeled by A (Directed Graph) • Gap edges for di- and tri-peptides • Potential sequence tag method (covered later)
Best Path of Spectrum Graph • How to find candidate paths • There are many paths, how to find the correct one? • We need scoring to evaluate paths
Find Candidate Paths • Heuristics: find a path with maximum number of edges • Longest path problem in DAG • DFS (Depth First Search)
Path Score • p(P,S) = probability that peptide P produces spectrum S= {s1,s2,…sq} • Scoring = computing probabilities
Finding Optimal Paths in the Spectrum Graph • For a given MS/MS spectrum S, find a peptide P’ maximizing p(P,S) over all possible peptides P: • Peptides = paths in the spectrum graph • P’ = the optimal path in the spectrum graph • Some software rank paths
Ratio Test Scoring for Partial Peptides • Incorporates premiums for observed ions and penalties for missing ions. • Example: for k=4, assume that for a partial peptide P’ we only see ions δ1,δ2,δ4. The score is calculated as:
Why Not Sequence De Novo? • De novo sequencing is still not very accurate! • Less than 30% of the peptides sequenced were completely correct!
W R V A L Database ofknown peptidesMDERHILNM, KLQWVCSDL, PTYWASDL, ENQIKRSACVM, TLACHGGEM, NGALPQWRT, HLLERTKMNVV, GGPASSDA, GGLITGMQSD, MQPLMNWE, ALKIIMNVRT, AVGELTK, HEWAILF, GHNLWAMNAC, GVFGSVLRA, EKLNKAATYIN.. Database ofknown peptidesMDERHILNM, KLQWVCSDL, PTYWASDL, ENQIKRSACVM, TLACHGGEM, NGALPQWRT, HLLERTKMNVV, GGPASSDA, GGLITGMQSD, MQPLMNWE, ALKIIMNVRT, AVGELTK, HEWAILF, GHNLWAMNAC, GVFGSVLRA, EKLNKAATYIN.. T G E P L K C W D T W R V A L T G E P L K C W D T De Novo vs. Database Search Database Search De Novo AVGELTK