SICKLE CELL ANEMIA
690 likes | 1.2k Vues
SICKLE CELL ANEMIA. JASON ABOUDI MOUABBI MD. What is the most common genetic disorder in the United States?. Outline. Background Inheritance Pathophysiology Diagnostic testing Prognosis Management of acute illness Long-term management Treatment options. What is a “Sickle”?.

SICKLE CELL ANEMIA
E N D
Presentation Transcript
SICKLE CELL ANEMIA JASON ABOUDI MOUABBI MD
What is the most common genetic disorder in the United States?
Outline • Background • Inheritance • Pathophysiology • Diagnostic testing • Prognosis • Management of acute illness • Long-term management • Treatment options
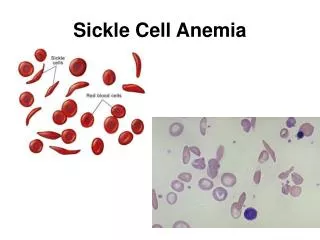
Normal vs Sickle Red Cells Sickle • Sickle-Shaped • Rigid • Lives for 20 days or less Normal • Disc-Shaped • Deformable • Life span of 120 days
Hemoglobin Genes and Products Hemoglobin is a tetramer of the product of 2 genes, 𝞪 globulin and non-𝞪 globulin There is 2 identical 𝞪 genes on Chr 16p 𝞫, 𝞭, 𝞬 genes are on chr 11p
Epidemiology Hemoglobin S Hemoglobin C Hemoglobin D Malarial Regions of Africa and Asia Hemoglobin E
SCD and Malaria Individuals with sickle cell trait have a milder course of P. falciparum infection Individuals with SCD-SS have more severe courses with a very high mortality rate
Sickle cell trait • Asymptomatic • Microscopic or gross hematuria (1-4%) • Recurrent • Persistent • Iron deficiency, renal obstruction, infection. • Medullary carcinoma of the kidney is more common in patients with SC. • Nephrectomy to control bleeding is almost never indicated because of the high probability of recurrence in the contralateral kidney. • With extreme exercise, hypoxia and dehydration, complications like splenic infarction and pain episodes may occur
Oxygenated state • Deoxygenated state
Diagnosis • Hemoglobin Electrophoresis • Peripheral blood smear
What is the most common cause of death in a 37 year old man with sickle cell disease? Cardiac disease Pulmonary disease Infections/sepsis Neurological Injury
The degree of hemolysis correlates with survival Mehdi Nouraie et al. Haematologica 2013;98:464-472
Vaso-occlusive Pain crisis • Early Signs • Fever • Tachycardia • Pallor • Late Signs • Pain in extremities • Back Pain • Abdominal Pain
Factors That Can Cause Sickle Cell Pain Crises • Infections • Low oxygen tension • Concomitant medical conditions (e.g., sarcoidosis, diabetes mellitus, herpes) • Dehydration • Acidosis • Extreme physical exercise • Physical or psychologic stress • Alcohol • Pregnancy • Cold weather
Management of sickle cell pain crisis • Management: • IV hydration: 1.5 x maintenance (not for acute chest) • Analgesics: • Acetaminophen • NSAIDs • Opiates • Oxygen to maintain O2 sat > 95% • Search for and treat the underlying cause
Question 25 year-old man with SCD presented with symptoms typical for acute pain crisis. Examination revealed BP of 14/72, HR of 72 an temp of 37. Mucus membranes are moist. Exam is unremarkable. In the ER, he received 1 mg of IV hydromophone and 1 L of 0.9% NS. What should you do next? Continue 0.9% NS Switch to 0.45 NS Switch to 3% NS Discontinue IVF
0.9 vs 0.45 Saline Choong K, Kho ME, Menon K, et al Hypotonic versus isotonic saline in hospitalised children: a systematic review Archives of Disease in Childhood 2006;91:828-835.
Question 38 year-old man presented with acute weakness of RUE. He has history of SCD-SS and takes folic acid daily. CBC showed WBC of 15,000, Hb of 9.4 and platelets of 400,000. What is the next step? Immediate exchange transfusion Immediate imaging of the brain Immediate administration of tPA Immediate administration of hydroxyurea
Question MRI showed acute arterial infarction as shown. What is the next step? Immediate exchange transfusion Immediate administration of tPA Immediate administration of hydroxyurea Physical therapy
Acute Chest Syndrome • New infiltrate • pleural effusion AND • Chest pain, cough, tachypnea, or wheezing OR • Fever > 38.5 • Drop in Hb(< 2g) and platelets (<175,000) Vichinsky et al, NEJM: 342;25. 1855-1865
Treatment • Treatment: • Analgesics • Broad-spectrum antibiotics • Bronchodilator and incentive spirometry • Simple or exchange transfusion • Simple: • when blood deoxygenation is not getting worse fast. • Exchange: • When O2 sat is 87 or lower and falling within 6 hrs. • When patient is confused (fat emboli). • When sternal pain is present (fat emboli) • Bronchoscopy is recommended in patients with no response to initial therapy
Acute Sickle Hepatic Crisis (ASHC) • Fever, RUQ abdominal pain, increase liver-associated enzymes and jaundice. • AST and ALT can go up in the 1000’s range • Bilirubin can be mildly elevated or severely elevated (up to 20 mg/dL) • Treatment: STAT and VIGOROUS exchange transfusion
Priapism • Persistent painful erection • Common among 5 and 10 boys and 20s and 50s in men • Treatment: ice packs, surgical ligation, intracavernous injection of alpha agonist (also PO), Surgical shunt, aspiration
Aplastic Crises • Parvovirus B19 • invades young erythroblasts in bone marrow • Presents with fever, URI • Progressive anemia with reticulocytopenia • Bone marrow recovery typically within 7-10 days Management: Transfusion if symptomatic with Hb drop.
Infections • By age 1, 30% of Hb SS pts are asplenic • By age 6, 90% are asplenic due to microinfarcts • Vulnerable to infection/sepsis with encapsulated organisms, • Strep pneumoniae (400x higher risk) • H. influenzae • Neisseria meningitidis • Osteomyelitis (Salmonella and Staph. aureus)
Acquired Hemosiderosis • Normal iron balance: 1mg/day • 1 unit of pRBC: ~250 mg • Acquired hemosiderosis is seen after 20 cumulative blood transfusions • Causes cardiac, hepatic impairment and endocrinopathy • Work-up: • ferritin (>1000) • MRI (liver and heart)
Acquired Hemosiderosis • Management: • Avoid unnecessary transfusion • Avoid consumption of iron • Use iron chelating therapy: • Deferoxamine IV • Deferasirox PO • Deferiprone PO
Long-term Management • Folic acid (chronic hemolysis) • Prophylactic antibiotics (children) • Vaccinations: • Pneumococcal • Meningococcal • H. influenza • Hepatitis A • Hepatitis B
Measures for Preventing Pain Crises in Patientswith Sickle Cell Disease • Consuming adequate amounts of fluids to prevent dehydration (especially during febrile periods and hot weather) • Avoiding exposure to extreme cold, exercising to exhaustion or using drugs that can lead to acidosis • Avoiding mountain climbing or air flights in an unpressurized cabin (noncommercial flights) above 10,000 feet • Avoiding hypoxemia in the perioperative period when general anesthesia is used or when a procedure involves hypertonic radiographic dyes
Increase HbF decreases HbS polymerization in RBC • Decreases sickling, crises, ACS • Decrease inflammatory cytokines • Lower WBC • Induces NOS-cGMP pathway in endothelium leading to a reduction of VTE • Decreases stroke incidence in CHILDREN (NOT Adults) Platt OS. N Engl J Med 2008;358:1362-1369.
Overall survival in patients with sickle cell disease who received hydroxyurea and in those who were treated conventionally James R. Eckman Blood 2010;115:2331-2332
Rivipansel (GMI-1070) Novel pan-selectin inhibitor being produced by Pfizer Telen MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood. 2015;125(17):2656-2664. doi:10.1182/blood-2014-06-583351.
L-glutamine Approved for Sickle Cell Disease • Studies found that the red blood cells affected by sickle cell disease are under high oxidative stress and try to protect themselves by producing a chemical called nicotinamide adenine dinucleotide (NAD) molecules. • The newly approved drug, L-glutamine, is an amino acid required to produce NAD molecules, and can therefore protect the red blood cells. • In a randomized clinical trial, sickle cell patients who took the medication had: • Fewer visits to the hospital for painkiller injections for painful episodes • Fewer hospitalizations • Reduced duration of hospitalizations • Fewer incidences of acute chest syndrome.
Pre-operative evaluation of sickle cell patients • Look for evidence of end-organ damage in respiratory, cardiovascular, neurological, liver and renal. • The minimal work-up should include completeblood count, electrolytes, renal function test, liver function test and chest x-ray. • I.V. fluid should requested during NPO period to avoid dehydration. • Hbgshould be > 7 mg/dl especially for major surgeries. • The intraoperative and postoperative periods can lead to acute severe sickling especially if the patient is exposed to dehydration, hypoxia, acidosis and low temperatures.