Download

1 / 24
300 likes | 708 Vues
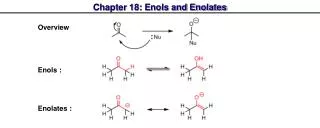
Titanium, Zinc and Zirconium Enolates. Metals such as Ti , Zn , and Zr give enolates which are intermediate in structural character between the largely ionic Li + enolates and covalent boron enolates.

E N D
Titanium, Zinc and Zirconium Enolates Metals such as Ti, Zn, and Zr give enolates which are intermediate in structural character between the largely ionic Li+ enolates and covalent boron enolates. The Ti, Zn, or Zr enolates provide oxygen-metal bonds that are largely covalent in character but can also accommodate additional ligands at the metal. Depending on the degree of substitution, both cyclic and acyclic transition states can be involved. • Titanium enolates can be prepared from lithium enolates by reaction with trialcoxytitanium(IV) chlorides, such as (isopropoxy)titanium chloride. Titanium enolates can also be prepared directly from ketones by reaction with TiCl4 and a tertiary amine.
Under these conditions, the Z-enolate is formed and the aldol adducts have syn stereochemistry. The addition can proceed through a cyclic transition state assembled around titanium. Titanium enolates can also be prepared from N-acyloxazolidinones. These enolates are considered to be chelated with the oxazolidinone carbonyl oxygen.
Trialkoxytitanium chlorides, which are somewhat less reactive, can also be used. Reactions of these enolates with aldehydes give mainly syn products, with the absolute stereochemistry being determined by the configuration of the oxazolidinone. These results are explained on the basis of a transition state which is hexacoordinate at titanium. The oxazolidinone substituent dictates the approach of the aldehyde. These reactions have been developed also in catalytic version.
Tin enolates can be generated from ketones and Sn(O3SCF3)2in the presence of tertiary amine. The subsequent aldol addition is syn-selective. Ph Tin(II) enolates prepared in this way also show good reactivity toward ketones as the carbonyl component.
N-Acylthiazolinethiones are also useful enolate precursors under these conditions. Uncatalyzed additions of trialkylstannyl enolates to benzaldehyde show anti stereoselectivity, suggesting a cyclic transition state.
Isolated tributylstannyl enolates react with benzaldehyde under the influence of metal salts including Pd(O3SCF3)2, Zn(O3SCF3)2, and Cu(O3SCF3)2 The anti : syn ratio depends on the catalyst. Zirconium enolates are prepared by reaction of lithium enolates with (Cp)2ZrCl2 (Cp = h5-C5H5). They act as nucleophiles in aldol addition reactions.
A comprehensive comparison of the anti : syn diastereoselectivity of the lithium, dibutylboron, and (Cp)2Zr enolates of 3-methyl-2-pentanone with benzaldehyde has been reported. E-enolate syn:antiZ-enolate syn:anti Li 17:83 Li 45:55 Bu2B 3:97 Bu2B 94:6 (Cp)2ZrCl 9:91 (Cp)2ZrCl 86:14 The order of stereoselectivity is Bu2B > (Cp)2Zr > Li These results are consistent with reactions proceeding through a cyclic transition state.
The Mukaiyama Reaction The Mukaiyama reaction refers to Lewis acid catalyzed aldol addition reactions of enol derivatives. The initial examples involved silyl enol ethers that do not react with aldehydes because they are a not strong enough nucleophile. However, Lewis acids do cause reaction to occur by activating the ketone. The simplest mechanistic formulation of the Lewis acid catalysis is that complexation occurs at the carbonyl oxygen, activating the carbonyl group to nucleophilic attack. If there is no other interaction, such a reaction should proceed through an acyclic transition state, and steric factors should determine the amount of syn versus anti addition.
This seems to be the case with BF3, where stereoselectivity increases with the steric bulk of the silyl enol ether substituent R'.
a-Substituted aldehydes show a preference for a syn relationship between the a substituent and hydroxy group. This is consistent with a Felkin-Ahn transition state. The results suggest that competition between antiperiplanar and synclinal transitions states is controlled by both steric and electrostatic effects.
The analysis of the transition-state effect on stereoselectivity has been extended to incorporate a,b-disubstituted systems. • Quite a number of Lewis acids besides TiCl4 and BF3 can catalyze the Mukaiyama reaction, including Bu2Sn(O3SCF3)2, Bu3SnClO4, Sn(O3SCF3)2, Sn(O3SCF3)2 and LiClO4. Triaryl perchlorate salts are also very active catalysts. • Trialkylsilyl cations may play a key role in Lewis acid-catalyzed reactions. Trimethylsilyl triflate itself is not a good catalyst, but in combination with other Lewis acids it generates excellent catalytic activity. Silyl enol ethers react with formaldehyde and benzaldehyde in water-THF mixtures with the use of lanthanide triflates such as Yb(O3SCF3)3 as catalysts. The catalysis reflects the strong affinity of lanthanides for carbonyl oxygen, even in aqueous solution.
It has been proposed that there may be a single-electron-transfer mechanism for the Mukaiyama reaction. • For example, photolysis of benzaldehyde dimethylacetal and 1-trimethylsilyloxycyclohexene in the presence of a typical photoelectron acceptor, triphenylpyrylium cation, gives an excellent yield of the addition product. • These reactions may operate by providing a source of trimethylsilyl cations, which act as the active catalyst.
Control of Enantioselectivity The most important factors in determining the syn or anti stereoselectivity of aldol and Mukaiyama reactions were identified as • the nature of the transition state (cyclic versus acyclic) and • the configuration (E or Z) of the enolate. Additional factors affect the enantioselectivity of aldol additions and related reactions. • Nearby chiral centers in either the carbonyl compound or the enolate can impose facial selectivity. • Chiral auxiliaries can achieve the same effect. • Finally, use of chiral Lewis acids as catalysts can also achieve enantioselectivity.
the nature of the transition state (cyclic vs acyclic) the syn or anti stereoselectivity the configuration of the enolate (E or Z) Nearby chiral centers and chiral auxiliaries in either the carbonyl compound or the enolate (facial selectivity) enantioselectivity chiral Lewis acids as catalysts (enantioselectivity) Control of Enantioselectivity Aldol and Mukaiyama reactions
Similarly, there will be a degree of selectivity between the two faces of the enolate when the enolate contains a chiral center. If both the aldehyde and enolate are chiral, mutual combinations of stereoselectivity will come into play. Up to this point, we have considered primarily the effect of enolate geometry on the stereochemistry of the aldol condensation and have considered achiral or racemic aldehydes and enolates. If the aldehyde is chiral, particularly when the chiral center is adjacent to the carbonyl group, the selection between the two diastereotopicfaces of the carbonyl group will influence the stereochemical outcome of the reaction.
One combination should provide complementary, reinforcing stereoselection… + = whereas the alternative combination would result in opposing preferences and lead to diminished overall stereoselectivity. ? + =
favored favored favored R-enolate R-aldehyde R-enolate S-aldehyde favored S-enolate S-aldehyde S-enolate R-aldehyde • The combined interactions of chiral centers in both the aldehyde and the enolate determine the stereoselectivity. The result is called double stereodifferentiation. OR
The analysis and prediction of the direction of preferred reaction depends on the same principles as for simple diastereoselectivity and are done by analysis of the attractive and repulsive interactions in the presumed transition state. • The analysis of results for a-substituted aldehydes with E- and Z-enolates indicates that the cyclic transition states shown below are favored with lithium and boron enolates. The larger a-substituent is aligned anti to the approaching enolate.
Complementary selectivity Ratio = 9 : 1 Opposed selectivity Ratio = 3.3 : 1 The stereoselectivity resulting from interactions of chiral aldehydes and enolates has been useful in the construction of systems with several contiguous chiral centers. major minor major minor
Other structural features may influence the stereoselectivity of aldol condensations. • One such factor is chelation by a donor substituent. Several b-alkoxyaldehydes show a preference for syn-aldol products on reaction with Z-enolates. • A chelated transition state can account for the observed stereochemistry. The chelated aldehyde is most easily approached from the face opposite the methyl and R' substituents. • A similar stereoselectivity has been noted for the TiCl4 mediated condensation of b-alkoxyaldehydes with silyl enol ethers.
The preceding reactions illustrate control of stereochemistry by aldehyde substituents. Substantial effort has also been devoted to use of chiral auxiliaries and chiral catalysts to effect enantioselective aldol reactions. • A very useful approach for enantioselective aldol condensations has been based on these oxazolidinones, which are readily available in enantiomerically pure form. • These compounds can be acylated and converted to the lithium or boron enolates by the same methods applicable to ketones. The enolates are the Z-stereo isomers.
The oxazolinone substituents R' then direct the approach of the aldehyde. Because of the differing steric encumbrance provided by the different oxazolidinones, the products have the opposite configuration at the new stereogenic sites. The acyl oxazolidinones are easily solvolyzed in water or alcohols to give the enantiomeric b-hydroxy acid or ester.
Enantioselectivity can also be induced by use of chiral boronates in the preparation of boron enolates. • Both the (+) and (-) enantiomers of diisopinocamphylboron triflate have been used to generate syn adducts through a cyclic transition state. The enantioselectivity was greater than 80% for most cases that were examined. • Another promising boron enolate is derived from (-)-menthone. It gives E-boron enolates that give good enantioselectivity and result in formation of anti products.
The stereoselectivity in these cases has its origin in steric effects of the boron substituents. Several hetereocyclic boron enolates with chirality installed at boron have been found to be useful for enantioselective additions. This diazaboridine is an example. • An additional feature of these chiral auxiliaries is the ability to select for syn or anti products, depending upon choice of reagents and reaction conditions. The diastereoselectivity is determined by whether the E- or Z-enolate is formed. • Considerable effort has been devoted to finding Lewis acid or other catalysts that could induce high enantioselectivity in the Mukaiyama reaction. As with aldol addition reactions involving enolates, high diastereoselectivity and enantioselectivity requires involvement of a transition state with substantial facial selectivity with respect to the electrophilic reactant and a preferred orientation of the nucleophile.