Download

1 / 77
770 likes | 941 Vues
Algorithms Exploiting the Chain Structure of Proteins. Itay Lotan Computer Science. Proteins 101. Involved in all functions of our body: metabolism, motion, defense, etc. Michael Levitt. Protein representation. Torsion angle model: C α model:. Structure determination.

E N D
Algorithms Exploiting the Chain Structure of Proteins Itay Lotan Computer Science
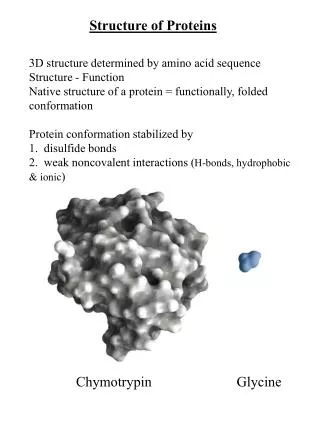
Proteins 101 Involved in all functions of our body: metabolism, motion, defense, etc. Michael Levitt
Protein representation • Torsion angle model: • Cα model:
Structure determination X-ray crystallography Bernhard Rupp
Outline • Fast energy computation during Monte Carlo simulation • Model completion for protein X-ray crystallography • Large scale computation of similarity Exploit specific properties of proteins to perform the computation efficiently
Outline • Fast energy computation during Monte Carlo simulation • Model completion for protein X-ray crystallography • Large scale computation of similarity Lotan, Schwarzer, Halperin* and Latombe. J. Comput. Bio. 2004 (to appear) *CS Department, Tel-Aviv University
Monte Carlo simulation (MCS) • Estimate thermodynamic quantities • Search for low-energy conformations and the folded structure Popular method for sampling the conformation space of proteins:
MCS: How it works • Propose random change in conformation • Compute energy E of new conformation • Accept with probability: Requires >>106 steps to sample adequately
Energy function • Bonded terms: • Bond lengths: • Bond angles: • Dihedral angles: • Non-bonded terms: • Van der Waals: • Electrostatic: • Heuristic: Go models, HP models, etc.
Pair-wise interactions • Cutoff distance (6 - 12Å) • Linear number of interactions contribute to energy (Halperin & Overmars ’98) Challenge: Find all interacting pairs without enumerating all pairs
Related work Biology • Neighbor lists • Verlet ’67 • Brooks et al. ’83 • Grid • Quentrec & Brot ’73 • Hockney et al. ’74 • Van Gunsteren et al. ’84 • Neighbor lists + grid • Yip & Elber ’89 • Petrella ’02 Computer Science • Bounding volume hierarchies for collision detection • Gotschalk et al. ’96 • Larsen et al. ’00 • Guibas et al. ’02 • Space partition methods for collision detection • Faverjon ’84 • Halperin & Overmars ’98 • Collisions detection for chains • Halperin et al. ’97 • Guibas et al. ’02
Grid method • Linear complexity • Optimal in worst case d:Cutoff distance
Contributions • Efficient maintenance and self-collision detection for kinematic chains • Efficient computation of pair-wise interactions in MCS of proteins • Scheme for caching and reusing partial energy sums during MCS • MCS software* Much faster than existing algorithm (grid method) *Download at: http://robotics.stanford.edu/~itayl/mcs
Properties of kinematic chains • Small changes large effects
Properties of kinematic chains • Small changes large effects
Properties of kinematic chains • Small changes large effects • Local changes global effects
Properties of kinematic chains • Small changes large effects • Local changes global effects • Few DoF changes long rigid sub-chains
Properties of kinematic chains • Small changes large effects • Local changes global effects • Few DoF changes long rigid sub-chains
ChainTree: A tale of two hierarchies • Transform hierarchy: approximates kinematics of protein backbone at successive resolutions • Bounding volume hierarchy: approximates geometry of protein at successive resolutions
TAI TAE TEI TCE TEG TGI TAC TAB TBC TCD TDE TEF TFG TGH THI D C G H A B E F I Hierarchy of transforms
BAH BEH BAD BCD BEF BGH BAB BB BA BC BD BE BF BG BH Hierarchy of bounding volumes
D C G H A B E F I The ChainTree TAIBAH TAEBAD TEIBEH TACBAB TCEBCD TEGBEF TGIBGH TABBA TBCBB TCDBC TDEBD TEFBE TFGBF TGHBG THIBH
D C G H A B E F I Updating the ChainTree TAIBAH TAEBAD TEIBEH TACBAB TCEBCD TEGBEF TGIBGH TABBA TBCBB TCDBC TDEBD TEFBE TFGBF TGHBG THIBH
P N O J K L M A B C D E F G H Computing the energy Recursively search ChainTree for interactions • Pruning rules: • Prune search when distance between bounding volumes is more than cutoff distance • Do not search inside rigid sub-chains
P N O J K L M A B C D E F G H Computing the energy [ ] P
P N O J K L M A B C D E F G H Computing the energy [ ] P [ ] N
P N O J K L M A B C D E F G H Computing the energy [ ] P [ ] [ ] N O
P N O J K L M A B C D E F G H Computing the energy [ ] P [ ] [ ] [ ] N N-O O
P N O J K L M A B C D E F G H Computing the energy [ ] P [ ] [ ] [ ] N N-O O [ ] [ ] [ ] J J-K K [ ] [ ] A-C C [ ] [ ] A-D C-D [ ] [ ] B-C D [ ] B-D
P N O J K L M A B C D E F G H [ ] A-C [ ] [ ] B-C D-G [ ] D-H Computing the energy [ ] P [ ] [ ] [ ] N N-O O [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] J J-K K J-L J-M K-L K-M L L-M M [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] A C A-E A-G C-E C-G E E-G H [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] A-B A-D C-D A-F A-H C-F C-H E-F E-H H-G [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] B D B-E B-G D-E F F-G G [ ] [ ] [ ] [ ] [ ] B-D B-F B-H D-F F-H
P N O J K L M A B C D E F G H [ ] A-C [ ] [ ] B-C D-G [ ] D-H Computing the energy E(O) [ ] P [ ] [ ] [ ] N N-O O [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] J J-K K J-L J-M K-L K-M L L-M M [ [ ] [ ] [ ] [ ] [ ] [ ] ] [ ] [ ] A C A-E A-G C-E C-G E E-G H [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] A-B A-D C-D A-F A-H C-F C-H E-F E-H H-G [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] B D B-E B-G D-E F F-G G [ ] [ ] [ ] [ ] [ ] B-D B-F B-H D-F F-H
Computing the energy • Only changed interactions are found • Reuse unaffected partial sums • Better performance for • Longer proteins • Fewer simultaneous changes
Computational complexity • Updating: • Searching: worst case bound Much faster in practice
Test 1-DoF change 5-DoF change [68 res.] [144 res.] [374 res.] [755 res.] [68 res.] [144 res.] [374 res.] [755 res.]
Simulation of α-Synuclein • 140 res. protein implicated in Parkinson’s disease • Multi-canonical Replica-exchange MC regime • Over 1000 CPU days of simulation • Study conformations at room temp. • Joint work with Vijay Pande
Outline • Fast energy computation during Monte Carlo simulation • Model completion for protein X-ray crystallography • Large scale computation of similarity Lotan, van den Bedem*, Deacon* and Latombe, WAFR 2004 van den Bedem*, Lotan, Latombe and Deacon*, submitted to Acta. Cryst. D * Joint Center for Structural Genomics (JCSG) at SSRL
Protein Structure Initiative • Reduce cost and time to determine protein structure 152K sequenced genes (30K/year) 25K determined structures (3.6K/year) • Develop software to automatically interpret the electron density map (EDM)
EDM 3-D “image” of atomic structure • High value (electron density) at atom centers • Density falls off exponentially away from center
Automated model building • ~90% built at high resolution (2Å) • ~66% built at medium to low resolution (2.5 – 2.8Å) • Gaps left at noisy areas in EDM (blurred density) Gaps need to be resolved manually
The Fragment completion problem • Input • EDM • Partially resolved structure • 2 Anchor residues • Length of missing fragment • Output • A small number of candidate structures for missing fragment A robotics inverse kinematics (IK) problem
Related work Biology/Crystallography • Exact IK solvers • Wedemeyer & Scheraga ’99 • Coutsias et al. ’04 • Optimization IK solvers • Fine et al. ’86 • Canutescu & Dunbrack Jr. ’03 • Ab-initio loop closure • Fiser et al. ’00 • Kolodny et al. ’03 • Database search loop closure • Jones & Thirup ’86 • Van Vlijman & Karplus ’97 • Semi-automatic tools • Jones & Kjeldgaard ’97 • Oldfield ’01 Computer Science • Exact IK solvers • Manocha & Canny ’94 • Manocha et al. ’95 • Optimization IK solvers • Wang & Chen ’91 • Redundant manipulators • Khatib ’87 • Burdick ’89 • Motion planning for closed loops • Han & Amato ’00 • Yakey et al. ’01 • Cortes et al. ’02, ’04
Contributions • Sampling of gap-closing fragments biased by the EDM • Refinement of fit to density without breaking closure • Fully automatic fragment completion software for X-ray Crystallography Novel application of a combination of inverse kinematics techniques
Two-stage IK method • Candidate generations: Optimize density fit while closing the gap • Refinement: Optimize closed fragments without breaking closure
Stage 1: candidate generation • Generate random conformation • Close using Cyclic Coordinate Descent (CCD) (Wang & Chen ’91, Canutescu & Dunbrack Jr. ’03)
Stage 1: candidate generation • Generate random conformation • Close using Cyclic Coordinate Descent (CCD) (Wang & Chen ’91, Canutescu & Dunbrack ’03)
Stage 1: candidate generation • Generate random conformation • Close using Cyclic Coordinate Descent (CCD) (Wang & Chen ’91, Canutescu & Dunbrack ’03)
Stage 1: candidate generation • Generate random conformation • Close using Cyclic Coordinate Descent (CCD) (Wang & Chen ’91, Canutescu & Dunbrack ’03)
Stage 1: candidate generation • Generate random conformation • Close using Cyclic Coordinate Descent (CCD) (Wang & Chen ’91, Canutescu & Dunbrack ’03) CCD moves biased toward high-density
Stage 2: refinement • Target function T(goodness of fit to EDM) • Minimize T while retaining closure • Closed conformations lie on Self-motion manifold of lower dimension 1-Dmanifold