Download

1 / 31
330 likes | 415 Vues
Learn about the intricate three-dimensional structure of proteins, including secondary and tertiary structures, denaturation, and folding processes. Understand the importance of weak noncovalent interactions in stabilizing protein conformations.

E N D
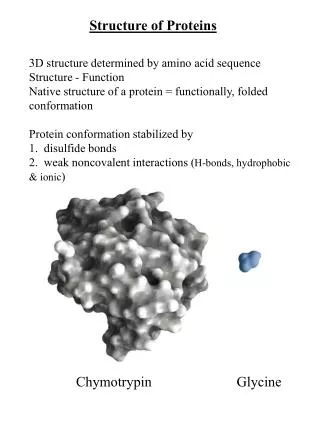
Chap. 4A The Three-dimensional Structure of Proteins • Topics • Overview of Protein Structure • Protein Secondary Structure • Protein Tertiary and Quaternary Structure • Protein Denaturation and Folding Fig. 4-1. Structure of the enzyme chymotrypsin, a globular protein
Overview of Protein Structure (I) In principle, a protein could have a nearly limitless number of shapes (structures) due to the fact that free rotation is allowed about many of its covalent bonds. However, the great majority of proteins have a specific chemical or structural function, which suggests that each has a unique 3D structure. This idea is supported by the finding that most proteins can be crystallized. Nonetheless, most proteins display at least a moderate degree of flexibility which is needed in their performance of function. Interestingly, parts of many proteins have no fixed structure. The lack of definable structure can be crucial to function.
Overview of Protein Structure (II) The spatial arrangement of atoms in a protein or any part of a protein is called its conformation. The functional conformation of the protein is called its native state. The native state is usually the conformation that is thermodynamically the most stable. A protein’s conformation is stabilized largely by multiple contributing weak noncovalent interactions. These include hydrogen bonds, ionic interactions, van der Waals interactions, and the hydrophobic effect of burying most nonpolar amino acid side-chains in the interior of the protein. Disulfide bonds also contribute to structural stabilization. In the native state, the number of weak noncovalent interactions is maximal.
Peptide Bond Structure X-ray diffraction studies of peptide bond structure by Linus Pauling and Robert Corey revealed that the C-N peptide bond is shorter than the C-N bonds in simple amines. Furthermore, the six atoms that are part of the peptide group of a peptide bond are coplanar. These data indicate that a resonance or partial sharing of two electron pairs occurs between the carbonyl oxygen and amide nitrogen (Fig. 4-2a). Because peptide bonds have partial double bond character, they do not rotate freely.
The Backbone of a Polypeptide Chain Rotation is permitted about the N-C (phi, ) and C-C (psi, ) bonds in the peptide backbone (Fig. 4-2b). Thus, the backbone of a polypeptide chain can be pictured as a series of rigid planes with consecutive planes sharing a common point of rotation at C. The rigid peptide bonds limit the range of conformations possible for a polypeptide chain. Note that the peptide bond occurs in a trans configuration 99.6 % of the time. Cis peptide bonds are very rare.
Ramachandran Plots of and Angles Both the and angles are defined as being ± 180˚ when the polypeptide backbone is fully extended and all peptide groups are in the same plane (Figs. 2c & d). In principle, and can vary freely between +180˚ and -180˚, but many values are prohibited by steric interference between atoms in the polypeptide backbone and amino acid side-chains. The conformation in which both and are 0˚ is prohibited for this reason. This conformation only serves as a reference point for describing the dihedral angles between peptide groups (planes). Allowed angles for and are shown in a Ramachandran plot for poly L-alanine (Fig. 4-3).
Overview of Protein Secondary Structure Secondary structure refers to stable, short-range, periodic folding elements that are common in proteins. A regular secondary structure occurs when each dihedral angle, and , remains the same or nearly the same throughout the element. There are a few types of secondary structures that occur widely in proteins. These include the a helix, the b conformation, and ß turns.
The helix (I) In the a helix (Fig. 4-4a), the peptide backbone adopts a cylindrical spiral structure in which there are 3.6 amino acids per turn (5.4 Å). The R groups point out from the helix axis, and mediate contacts to other structure elements in the folded protein. The helix is stabilized by hydrogen bonds between backbone carbonyl oxygen and amide nitrogen atoms that are oriented parallel to the helix axis. In fact the structure maximizes the use of internal hydrogen bonding. Hydrogen bonds occur between residues located in the n and n + 4 positions relative to one another. The helix forms more readily than many other conformations in part because of its optimal use of internal hydrogen bonding.
The helix (II) Other views of the helix are shown in Figs. 4-4 b-d. Fig. 4-4 b shows an end-on view of an helix, and emphasizes how R groups are located on the surface. A space filling model of an helix is shown is Fig. 4-4 c, and shows how there actually is no central hole in the helix as seems to be the case using the ball-and-stick model in Fig. 4-4 b. Fig. 4-4 d shows a helical wheel projection of an helix and emphasizes how the side chains of amino acids in the n and n + 3 positions are in close contact. It also emphasizes how different faces of the helix can have different properties, e.g., polarity.
and Angles for Secondary Structures The backbone atoms of amino acid residues in the prototypical helix have characteristic dihedral angles that define the helix conformation (Table 4-1). helical segments in proteins often have dihedral angles that deviate slightly from these ideal values. This introduces slight bends and kinks into the helix axis.
Helix Nomenclature Protein structure studies have determined that right-handed helices occur in proteins (see figure in Box 4-1). Extended left-handed helices have not been observed in proteins, presumably because they are theoretically less stable. Experiments have shown that an helix can form in a polypeptide consisting of either L- or D-amino acids. However, all residues must be of one stereoisomeric form or the other or the helix will be disrupted. The most stable form of an helix formed by D-amino acids is left-handed.
Worked Example 4-1. Secondary Structure and Protein Dimensions
Helix Stability and Amino Acid Composition Five types of constraints affect the stability of an helix: 1) the intrinsic propensity of an amino acid residue to form an helix (Table 4-2, next slide); 2) the interactions between R groups, particularly those spaced three or four residues apart; 3) the bulkiness of adjacent R groups; 4) the occurrence of proline and glycine residues; and 5) interactions between amino acid residues at the ends of a helical segment and the electric dipole inherent to the helix (Fig. 4-5, below).
Helical Preferences of Amino Acids The properties of the R group strongly affect the capacity of the backbone atoms to take up the characteristic and angles of an helix (Table 4-2). Alanine with its small methyl group in its side chain shows the greatest propensity to form an helix under most conditions. In contrast, amino acids such as threonine and asparagine, with bulky groups attached to the ß carbon of the amino acid show a reduced propensity to occur within an helix. Proline and glycine show little tendency to occur in helices. In proline the N atom is part of a rigid ring, and rotation about the N-C bond is not possible. This places a destabilizing kink into an helix. In addition, the N atom when in a peptide bond linkage has no substituent hydrogen that can participate in hydrogen bonding to other residues. Glycine is highly conformationally flexible due to its having a H atom for a side chain. This is disruptive to the stability of the helix.
Helix Dipoles and Helix Stabilization A small electrical dipole exists in each peptide bond. These dipoles are aligned through the hydrogen bonds of the helix, resulting in a net dipole along the helical axis that increases with helix length (Fig. 4-5). For this reason, the helix is stabilized when amino acids with negatively charged side chains are located at the amino terminus of the helix, and vice versa. Placement of an amino acid with a positively charged side chain near the amino terminus of an helix destabilizes it, as does the placement of an amino acid with a negatively charged side chain near the carboxyl terminus of the helix.
ß Conformation A second type of secondary structure, the ß conformation, is very common in proteins. In this structure element, the polypeptide backbone is nearly fully extended into a zigzag strand rather than a helical structure (Fig. 4-6 a). The R groups of consecutive amino acids in a ß strand are oriented on opposite sides of the strand. As expected, the and angles for the ß conformation are distinctly different from those observed in the helix (Table 4-1).
Antiparallel and Parallel ß Sheets The arrangement of several ß strands side-by-side forms a planar type structure called a ß sheet (a.k.a., ß pleated sheet) (Fig. 4-6 b & c). Hydrogen bonds between adjacent strands of the sheet stabilize the structure. The adjacent strands in a ß sheet can be either parallel or antiparallel, that is, having the same or opposite N-to-C terminal orientations, respectively . The hydrogen-bonding patterns are slightly different for the antiparallel and parallel ß sheets, with hydrogen bonds being more perfectly aligned in the former.
Structures of ß Turns ß turns are common secondary structure elements that link successive runs of helix or ß conformation where the polypeptide chain reverses direction in space (Fig. 4-7). ß turns therefore, commonly are located at the surface of a globular protein. Particularly common are ß turns that connect the ends of two adjacent strands of an antiparallel ß sheet. The most common types of ß turns (Type I and Type II ß turns) contain four amino acids in which the first and fourth residues in the turn are hydrogen bonded to one another. The second and third residues in the ß turn commonly hydrogen bond to water at the surface of the protein.
Proline Isomers Glycine and proline often are present in ß turns. Glycine is prevalent because its side chain is small, making the peptide backbone quite flexible. Proline is common because peptide bonds involving the imino nitrogen of proline readily assume the cis configuration (Fig. 4-8). For peptide bonds involving the imino nitrogen of proline, about 6% are in the cis configuration and many of these occur in ß turns.
Ramachandran Plots of Secondary Structures The idealized dihedral angles that define the helix and ß conformation fall within relatively restricted regions of sterically allowed structures on a Ramachandran plot (Fig. 4-9a). Most experimentally determined values of and measured in known protein structures fall into these regions, with high concentrations near the helix and ß conformation values as predicted (Fig. 4-9b).
Overview of Tertiary and Quaternary Structure Tertiary structure refers to the overall three-dimensional arrangement of all atoms in a protein. Tertiary structure deals with long-range aspects of the fold of a protein, including interactions that form between isolated elements of secondary structure. Both noncovalent and covalent interactions are included in the tertiary structure. Quaternary structure refers to the contacts between, and overall arrangement in three-dimensional space of the individual subunits of a multisubunit protein.
Fibrous and Globular Proteins Many proteins can be classified into two groups based on their general structural features. Fibrous proteins, such as -keratins and collagens, have polypeptide chains arranged in long strands or sheets. These proteins usually consist mostly of a single type of secondary structure and their tertiary structure is relatively simple. Globular proteins, such as myoglobin and serum albumin, consist of polypeptide chains that are folded into a spherical or globular shape. Globular proteins often contain several types of secondary structure. Fibrous proteins generally function to provide support, shape, and external protection to vertebrates; globular proteins often function as regulatory proteins and enzymes. The dimensions of globular proteins (e.g., human serum albumin, 585 aas) are much smaller than they would be if their chains were exclusively helical or adopted the ß conformation (Fig. 4-15).
Examples of Fibrous Proteins The properties of the fibrous proteins we will discuss are summarized in Table 4-3. All of these proteins are insoluble in water, and they contain a high proportion of nonpolar amino acid residues both in their interiors and on their surfaces. The hydrophobic surfaces are largely buried because many similar polypeptide chains are packed together to form elaborate supramolecular complexes.
Fibrous Proteins: -Keratins (I) The -keratins, which are only found in mammals, constitute most of the dry weight of hair, wool, nails, claws, quills, horns, hooves, and much of the outer layer of skin. Hair -keratin (Fig. 4-11) consists of an elongated right-handed helix with somewhat thicker structures near the N- and C-termini. Pairs of -keratin monomers interwind in a left-handed sense, forming two-chain coiled coils. These then combine in higher order structures called protofilaments, protofibrils, and intermediate filaments. Intermediate filaments contain 32 monomeric strands of -keratin. -keratin is rich in the hydrophobic amino acids Ala, Val, Leu, Met, and Phe. -keratins are strengthened by covalent disulfide bond cross-links between monomers in the coiled-coil and higher order structures. In rhinoceros horn -keratin, ~18% of the residues are cysteines involved in disulfide bonds.
Fibrous Proteins: -Keratins (II) The -keratins of hair lengthen when exposed to moist heat. This occurs due to penetration of water into the helices of hair fibers where it competes with intrachain hydrogen bonds. The hydrated, stretched -keratins adopt a more ß conformation-like structure. Hair -keratins are disulfide bond-linked, and consecutive reduction and oxidation steps are used in the process of waving and curling hair (permanents, figure in Box 4-2).
Fibrous Proteins: Collagen Collagen is a structural protein found in connective tissue such as tendons, cartilage, the organic matrix of bone, and the cornea of the eye. The collagen helix has a unique secondary structure which is left-handed and has three amino acids per turn (Fig. 4-12 a & b). Three collagen polypeptides ( chains) associate in a right-handed superhelical coiled coil structure called the three-stranded collagen superhelix (Fig. 4-12c & d). Vertebrate collagen typically contains about 35% Gly, 11% Ala, and 21% Pro and 4-hydroxyproline. The Pro and 4-Hyp residues permit the sharp twisting of the collagen helix. The repeating tripeptide Gly-X-Y, where X is often Pro and Y is often 4-Hyp, occurs frequently in the chain sequence. Gly residues are located where the three chains of the collagen superhelix contact one another (Fig. 4-12 d, red).
Structure of Collagen Fibrils (I) Collagen (Mr 300,000) is a rod-shaped molecule about 3,000 Å long and only 15 Å thick. Its three helically intertwined chains can have different sequences and each chain has about 1,000 amino acid residues. Collagen fibrils are made up of collagen molecules aligned in a staggered fashion and cross-linked for strength. The specific alignment and degree of cross-linking vary with the tissue and produce characteristic cross-striations in an electron micrograph (Fig. 4-13, upper). A typical mammal expresses on the order of 30 structural variants of collagen. Diseases such as osteogenesis imperfecta and Ehlers-Danlos syndrome are caused by mutant alleles of collagen genes. Commonly an amino acid with a relatively large R group such as Cys or Ser replaces Gly residues in mutant collagens, disrupting their structure and function.
Structure of Collagen Fibrils (II) The individual chains of triple-helical collagen molecules and the collagen molecules of fibrils are cross-linked by covalent bonds involving lysine, 5-hydroxylysine, and histidine residues that are present at some of the X and Y sites in the triplet repeat. The dehydrohydroxylysinonorleucine residue produced by cross-linking between lysine and 5-hydroxylysine residues is shown below. With aging, cross-linking frequency increases contributing to the brittle properties of aging connective tissue.
Synthesis of 4-hydroxyproline As noted above,collagen chains are constructed of the repeating tripeptide unit Gly-X-Y, where X often is Pro and Y often is 4-Hyp. The proline ring normally occurs as a mixture of two puckered conformations, called C-endo and C-exo (see below). The collagen helix requires that the Pro residue in the Y positions of the repeat adopt the C-exo conformation. This conformation is enforced by the 4-hydroxyl group of 4-Hyp, and this is why this amino acid occurs in collagen chains. In the absence of vitamin C, cells cannot hydroxylate the Pro in the Y positions. This leads to collagen instability and the connective tissue problems seen in the deficiency disease known as scurvy. 4-Hyp is synthesized by the enzyme prolyl 4-hydroxylase.
Vitamin C and Scurvy (I) Vitamin C (L-ascorbic acid) plays an important role in the production of collagen. In vitamin C deficiency (scurvy), connective tissue synthesis and function are impaired (Box 4-3). In extreme deficiency cases, the individual experiences numerous small hemorrhages caused by fragile blood vessels, tooth loss, poor wound healing and the reopening of old wounds, bone pain and degeneration, and eventually heart failure. Vitamin C is important in maintaining the enzyme prolyl 4-hydroxylase in its active state. In the normal prolyl 4-hydroxylase reaction (see below), one molecule of -ketoglutarate and one O2 bind the enzyme along with a Pro residue. The -ketoglutarate is oxidatively decarboxylated to form succinate and CO2, and the remaining oxygen atom is used to hydroxylate the appropriate Pro residue in procollagen. L-ascorbate is not needed in this reaction.
Vitamin C and Scurvy (II) L-ascorbic acid plays a vital role in preventing prolyl 4-hydroxylase from being inactivated in a second reaction that it commonly catalyzes. In this reaction (see below), -ketoglutarate is oxidatively decarboxylated to succinate and CO2 in a reaction that does not involve hydroxylation of proline. In this reaction a critical Fe2+ metal ion of the enzyme is oxidized to the Fe3+ state, inactivating the enzyme for further catalysis. When L-ascorbic acid is present, the iron atom is reduced to the Fe2+ state, maintaining the active form of the enzyme.