The Main MOE Windows
The Main MOE Windows. MOE Database Viewer ( DBV ) Cheminformatics, conformational search, fingerprints, clustering, combinatorial library design. The MOE Window ( MOE ) or ( ) Small molecule bioinformatics, Molecular mechanics, Small molecule visualization, Forcefield applications.
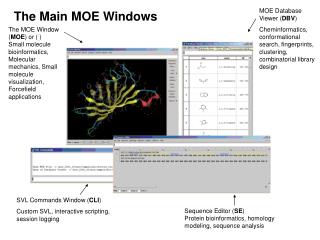

The Main MOE Windows
E N D
Presentation Transcript
The Main MOE Windows MOE Database Viewer (DBV) Cheminformatics, conformational search, fingerprints, clustering, combinatorial library design The MOE Window (MOE) or ( ) Small molecule bioinformatics, Molecular mechanics, Small molecule visualization, Forcefield applications SVL Commands Window (CLI) Custom SVL, interactive scripting, session logging Sequence Editor (SE) Protein bioinformatics, homology modeling, sequence analysis
Intro 1: The MOE Window • Used for: • Building small molecules • Molecular mechanics • Structure-based drug • design • Docking • SCF Calculations • Molecular dynamics • Flexible alignment • PH4 elucidation • Conformational searching
Intro 2: The Sequence Editor • Used for: • Protein bioinformatics • Sequence alignment • Homology searching • Homology modeling • Target family analysis • RCSB download • Consensus modeling • PDB searching
Intro 3: The MOE Database Viewer • Used for: • Cheminformatics • QSAR • Conformation search output • Dynamics output • Flexible alignment output • Docking output • Clustering • Fingerprints • Similarity search • Diverse subset • Data correlation • Combinatorial library design • R-group preparation • PH4 searching • Washing / Preprocessing
Intro 4: The SVL Commands Window Used for: • Custom SVL • Interactive scripting • Session logging
Layout of Course • Main MOE Window: • Opening/saving files • Building molecules • Rendering • Introduction to the Sequence Editor • Introduction to the Database Viewer • Basics of molecular mechanics and conformational searching • Basics of cheminformatic analysis with the Database Viewer • Comments on SVL
1. Structure of the MOE Window Task Cancel Button Main Menu Commands RHS Button Bar SVL Command Line Popup menu 3D Rendering Area Footer Pager Bar
MOE Menu Command Conventions • Commands from the MOE Window are preceded by MOE or () (Render | Backbone | Color | Chain Color) or (MOE | Render | Backbone | Color | Chain Color)
Mouse Conventions in MOE General Mouse Actions 3-Button Mouse – 2-Button Mouse Mapping LEFT - Selecting objects, menu commands MIDDLE - Rotating, translating moving objects Press and release <Alt> key RIGHT - SE and DBV Popup menus
Input / Output File Formats in MOE • MOE can read various input formats, e.g. MOE, PDB, SD etc. • A variety of export file formats are possible, e.g. MOE, Tripos MOL2 etc. • Picture files may be generated for publications or presentations.
Opening Files in MOE – (File | Open) Current Path/ Directory Change Working Directory (CWD) Path Text Field Enforce File Type ‘..’ go up a Directory Operations to Perform on selected file(s) Recent Directories List Directory/ File List Open file in text editor Open file into MOE
Exercise: Opening a File • Open the File Open panel (File | Open). • Use the pull-down menu to switch to the $MOE/sample/mol directory. • Find the file $MOE/sample/mol/sulph_quin.moe • Open MOE file into the MOE Window by either: • Selecting the file and clicking OK or Open in MOE. • Double left-clicking on the filename.
Exercise: Opening a File (cont.) • Center the View (Render | View or RHS | View). • Render the molecule in stick mode (Render | Stick).
Exercise: Manipulate molecule in 3D Window Right Click: Popup menu Middle click: Change center of rotation Left Click: Select atoms one at a time Left Click in Empty Space: To de-select to clear Middle Drag: XY Rotate Ctrl Middle Drag or Scroll wheel Zoom in/out Left Drag: Selection Box
Exercise: Render molecule • Render as ball and stick, label all atoms by name, and show H-Bonds • MOE | Render | Draw | Hydrogen Bonds • Mode | Ball and Stick 3. Label | Name 4. Label | Clear
Exercise: Saving a picture (1) 1. Choose MOE | File | Save 2. Click on MkDir to create a new directory called ‘course’ 3. Click on Set CWD to set as the new working directory
Exercise: Saving a picture (2) • Choose Save ‘Picture’ • Enter the filename ‘sulph_quin.png’ • Choose Format ‘PNG’ • Finally click on Save
Rendering 2D Depicted Molecules 1. MOE | Edit | Automatic | Depict as 2D • Press ‘Export Bitmap’ to save picture as ‘sulph_quin2.png’ • Press OK • Press Close
Drawing Molecular Surfaces • Create Molecular Surfaces via the Molecular Surface panel • (MOE | Compute | Surfaces and Maps) • Manage surfaces and other graphics objects with the Graphic Object Manager • (MOE | Window | Graphic Objects)
Molecular Surfaces and Maps • Tool for active site analysis • Integration of three applications:Molecular Surfaces, Contact Preference Maps and new Electrostatic Maps • Easy control of definition for atom sets • Automatic handling of surface names for easy comparisons • Build molecular surfaces • Gaussian, Connolly and VDW • Color by various properties • Predict contact preferences • Plot knowledge based potentials for hydrophilic and hydrophobic contacts • Calculate electrostatic maps • Plot electrostatically preferred positive, negative and neutral regions
Exercise: Drawing Molecular Surfaces (1) • Draw a surface around the inhibitor by first choosing (MOE | Compute | Surfaces and Maps) using the default options • Press Apply
Exercise: Outputting the system… to the printer To print the current MOE 3D window choose MOE | File | Print... Printer or Postscript file Landscape/Portrait Header Footer
Exercise: Outputting the system… to various formats • Save the current MOE 3D window. MOE | File | Save... 2. Enter filename to save ‘my_sulph_quin.moe’ • Choose Save: Molecule and Format: .moe. To save surface, select Graphics:All 4. Click on Save
Exercise: Close System and Open Builder • Close the current system (MOE | RHS | Close) 2. Open the Builder (MOE | RHS | Builder)
The Molecule Builder Other atom types, including dummy atom at centroid Edit or Add Element Edit Ionization State Edit Chirality Fragment substitution buttons Enter Fragment SMILES string • Edit: • Compound Name • Bond Length • Bond Angle • Torsion Library of functional groups Undo button
Exercise: Build a molecule Press Select C Press Press Select C
Exercise: Build a molecule (cont.) Press Shift-select 2 C <Shift> Shift-select 2 C Press <Shift>
Exercise: Build a molecule (cont.) Select H Press Select H Press 4 times
Exercise: Build a molecule (cont.) Press Press Select H Press Deselect H
Exercise: Energy Minimize RHS | Minimize
Save Molecule • Save the current MOE 3D window as a MOE file. MOE | File | Save... Enter filename to save ‘my_first_molecule.moe’ Save ‘Molecule’ Choose Format ‘MOE’ 2. Press Save
Protein and Carbohydrate Builders MOE | Edit | Build | Protein or SE | Edit | Protein Builder MOE | Edit | Build | Carbohydrate
Selecting Atoms with the Left Mouse Button <Ctrl>-Left Click: Auto-extend selection to residue Left Click: Select atoms one at a time <Ctrl> <Shift> <Shift>-Left Click: Add to / toggle atom selection Note on <Ctrl>-Left Click: There is only one residue in the built molecule, so the whole molecule will be selected. This will be revisited later. Left Drag: Selection Box
Exercise: Selecting Atoms with the Left Mouse Button • Use Left-Click to select atoms one at a time. • Use Left-Drag to draw a selection box. • Use <Shift>-Left Click extend/toggle atom selections. • Use <Ctrl>-Left Click to select entire residues. • Left-mouse click in empty space to de-select any atoms.
Exercise: Fixing/Unfixing Atoms (Edit | Potential | Fix / Unfix) • Fixing atoms: • Left mouse drag to select the atoms to be fixed. • Fix the atoms with the command (Edit | Potential | Fix) . • Fixed Atoms do not move until unfixed. • Unfixing atoms: • Select the atoms to be unfixed. Use (Selection | Potential | Fix) to select all the fixed atoms. • Unfix the atoms with the command (Edit | Potential | Unfix). Once unfixed the atoms may move.
Exercise: Fixed atoms and rotatable bonds If two atoms in a rotatable bond are selected <Alt>-Left Drag will rotate about the bond. If no atoms are fixed, the small group rotates by default. <Alt> Drag The larger group can be forced to rotate by fixing an atom in the smaller group FIX this atom <Alt> Drag
Meters and Measurement (Edit | Measure) or (RHS | Measure…) • Choose Distances to measure and display the distance between two atoms. • Choose Angles to measure and display the angle between three atoms. • Choose Dihedrals to measure and display the dihedral angle between four atoms.
Meters – Creating and Removing To create a meter, choose MOE | Edit | Measure | Distances. To remove it, select the atoms involved, and use RHS | Remove | Distances.
CLI Prompt Menus One-line CLI Prompt menus occupy the SVL Command Line at the top of the window Press (Esc) to exit the prompts or choose to delete the process using the ‘Cancel’ button on the top right.
MOE Selection Menu Extend selection set Deselect all atoms Invert current selection Knowledge-based selectors for different parts of protein/ligand bound structures Atom Selection Tool for advanced selecting Pull down menus for selection by property, element, extension or other criteria Save and Restore selection sets Selection state of residues is coordinated with the Sequence Editor
The Atom Selector MOE | Selection | Atom Selector General Selection actions Selection Restrictions Logic Operations Save and Load selection sets; create named sets Select by name Select by elements and atom types Select by SMILES string substructure • Other Selection Options: • Accessibility • Chirality • Connectivity • Geometry • General • Pharmacophore • Protein (e.g. alpha carbon) Extend Selection Criteria
Moving Atoms with the Middle Mouse Button <Alt> Middle Drag: XY Rotate center of rotation Middle Drag: <Alt> <Shift>-Middle Drag: XY Rotate selected only Middle click to center on atom XY Translate selected only XY Translate <Shift>Middle Drag: Zoom in/out <Ctrl>Middle Drag:
Exercise: Moving Atoms with the Middle Mouse Button • View the coordinate system (Render | Draw | Coordinate Axes). • Rotate view about the XY axes (Middle Drag). • Translate view (<Shift>-Middle Drag). • Middle click on the carbonyl O to move the center of rotation. • Remove the coordinate axes by de-selecting (Render | Draw | Coordinate Axes) • Deselect all atoms (Left-click in space or (Selection | Clear)). • Move a selected subset with <Alt>-Middle Drag (rotate)<Shift><Alt>Middle Drag (translate).
MOE Render Menu Center, Save and Load views Draw H-bonds, VDW contacts, label options, coordinate axes, etc. Stereo viewing options: Quad-Buffer, Over-Under, Interlace, Left-Right, Parallel Protein/DNA backbone rendering Atomic/Molecule object rendering Hide and Show various sets Basic coloring Atom Labeling menu Detailed atom and label style menu Setup of default colors and object dimensions.
Exercise: Small Molecule Rendering • Rendering actions apply to: • All atoms (if none are selected) • Selected atoms only (if there are selected atoms) • Deselect all atoms. • Use Left drag click to select part of the molecule. Render it as space filling(Render | Space Filling). • Select other parts of the molecule using left-drag or other methods, and render them as stick, ball and stick, or line. Select and render as ‘Space Filling’ Select group and render as ‘Stick’
Protein Backbone Rendering MOE | Render | Backbone or MOE | Popup | Backbone Turn off backbone Various Backbone rendering styles Backbone coloring options
Exercise: Open PDB file prior to rendering complex • Close the current system: MOE | File | Close • Select the file MOE | File | Open$MOE/sample/mol/1pph.pdb. • Press ‘Load PDB File’. • A variety of options are available in the PDB File panel. Choose to centre the view and press ‘OK’.
Exercise: Rendering Trypsin with Ligand • Select the water molecules (Selection | Solvent) and delete them (RHS | Delete). • Select the ligand(s) (Selection | Ligand) and render as space filling (Render | Space Filling). • Use Render | Color to select a desired colour for the selected atoms (green). • Deselect the atoms • Draw a backbone ribbon through the selected atoms (Render | Backbone | Slab Ribbon). • Color the backbone by Chain Color(Render | Backbone | Color | Chain Color). • Hide the selected protein atoms(Render | Hide | Receptor). • Click on empty space to clear selection state.
Exercise: Protein-Ligand Pocket Rendering • Turn the backbone off (MOE|Popup|Backbone|None) • Show ligand and pocket (MOE|Popup|Show|Ligand, Pocket) • Label the residues (MOE | RHS | Label | Residue) • Draw H-bonds (Render | Draw | Hydrogen Bonds) • Center the image (RHS | View) which should now look like the image on the left. • Save the system as a MOE file, MOE | File | Save trypsin_pocket.moe
v Preference for Hydrophilic Contacts u P r Preference for Hydrophobic Contacts Contact Statistics Calculate and display probability of finding hydrophobic or hydrophilic contact at a point P relative to an atom. The contacts are derived from PDB x-ray structure statistics.