Download
1 / 30
300 likes | 491 Vues
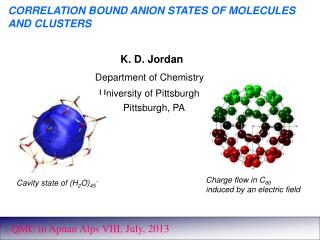
K. D. Jordan. Department of Chemistry. University of Pittsburgh Pittsburgh, PA. CORRELATION BOUND ANION STATES OF MOLECULES AND CLUSTERS. Charge flow in C 60 induced by an electric field. Cavity state of (H 2 O) 45 -. . QMC in Apuan Alps VIII, July, 2013. Support.

E N D
K. D. Jordan Department of Chemistry University of Pittsburgh Pittsburgh, PA CORRELATION BOUND ANION STATES OF MOLECULES AND CLUSTERS Charge flow in C60 induced by an electric field Cavity state of (H2O)45- . QMC in Apuan Alps VIII, July, 2013
Support Acknowledgements and Projects National Science Foundation Department of Energy Group members who contributed to the work in this area: F. Wang (U. Arkansas) T. Sommerfeld (Southeastern Lousiana Univ.) T.-H. Choi (Choongnam National Univ.) V. Voora Collaborators M. Johnson (Yale Univ.)
Reduction of CO2 using (H2O)n- clusters* Experiment starts with a cold cluster (T ~ 50K) with the e- localized on (H2O)6 Following vibrational excitation of either water (OH stretch or bend) or CO2 (asymm stretch) the electron jumps to the CO2 • Ab initio MD simulations reveal that there are a large number of different reaction pathways. • But in each case ET is triggered by formation of a H-bond to CO2 *J. Breen, A. F. DeBlase, T. L. Guasco, V. K. Voora, K. D. Jordan, T. Nagata and M. A. Johnson, J. Phys. Chem., 116, 903 (2012)
Classification of anion states • Unbound: temporary anions (resonances) • Bound • Bound at KT/HF level • Unbound at KT/HF level Pose some of the same problems as resonances • In this talk I focus on non-valence correlation-bound anions • E.g., certain cavity-bound anion states of (H2O)n clusters and the s-type anion state of C60 Model system comprised of four water molecules Electron binding energy (EBE) calculated vs. R R EBE = EEES + Econf + Edisp EES = exch. plus electrostatic; conf. = effect of confinement on KE; disp=dispersion Hartree-Fock: essentially the sum of the first two terms
Methods considered MP2 MP2 for anion and neutral CCSD(T) CCSD(T) for anion and neutral EOM-CCSD 1p + 2p1h CI for anion using transformed H EOM-MP2 1p + 2p1h CI for anion using transformed H ADC(2) second-order self energy with off-diagonal coupling Diag-ADC(2) second-order self energy without off-diagonal coupling OO-MP2 orbital-optimized MP2 for anion and neutral OO-CCD orbital-optimized coupled cluster doubles for anion and neutral B-CCD Bruekner orbital coupled cluster doubles for anion and neutral QMC quantum Monte Carlo Methods in blue: allow for relaxation of the singly occupied orbital in response to correlation effects
Results for a basis set with very diffuse functions: aug-cc-pVDZ+6s6p MP2 and coupled cluster methods fail when HF cease to bind the e-. Coupled cluster methods still useful as long as HF gives binding.
Results for basis set without highly diffuse functions: aug-cc-pVDZ In the absence of diffuse functions, even MP2 binds the e- near R = 4 A, because singly occ. orbital has appreciable weight in molecular region (fortuitous) Overall shape of the binding curve is not correct
Singly occ. NO from HF and EOM-CCSD natural orbital analysis (in latter case ~ Dyson orbital from Green's function treatment) • At large R, the anion is bound in the HF approx. • It ceases to bind around R = 4.2 Å • Turns into an approximate continuum function
ADC(2), OO-MP2, and B-CCD all give for (H2O)4 EBEs reasonably close to the EOM values even when using large flexible basis sets • Establishes that for binding of an e- to (H2O)n clusters, high-order correlation effects are not of major importance • More important is the relaxation of the singly-occupied orbital in response to the correlation effects • As we will see later, for many other systems, high-order correlation effects play a more significant role The ab initio results for the (H2O)n- clusters, have been used to test model potential approaches that we have been developing Our most sophisticated approach employs three mutually interacting, atom-centered polarizable sites per water, and allows for self-consistent treatment of e--water and water-water polarization
Consider a (H2O)24 (W24a) cluster • In addition consider W4, W8, W12, W16, and W20 cut out of W24. Surfaces that enclose 70% of the charge density of the excess electron (from pol. model ) 1V. Voora, T. Sommerfeld, K. Jordan, V. Vysotskiy, L. Cederbaum, JTCT, 2012
EBEs of W24a and subclusters extracted from W24a ADC refers to ADC(2) Green's function method. Pol1 decouples e- interacting with induced dipoles from water-water interactions and e- directly inducing dipoles. Pol3-SC treats these interactions self- consistently. C = large set of diffuse s + p functions at COM • For all clusters our self-consistent polarization model gives EBEs in excellent agreement with the ab initio results • Self-consistent treatment of e--water and water-polarization is essential for the cavity-type anion states
Any system with sufficient polarizability should support non-valence correlation-bound anions This includes species such as C60 • Evidence in electron-scattering and Rydberg atom collision experiments that C60 captures 0 eV electrons • One possible interpretation is the existence of an s-like polarization bound anion • Not identified in ab initio calculations carried out to date.
STM dI/dV images of so-called superatom states of a C60 monomer and dimer on the copper surface (Petek group)1 Dimer Monomer However, the existence of such an anion for a C60 adsorbed on a metal surface does not mean it would be bound in the gas phase (image potential stabilization) 1Feng et al. Science 320, 359 (2008)
One-particle energy levels of C60 Hg (-1.0 eV) Ag (?) Negatives of electron affinities T2u (-1.4 eV T1g (-2.0 eV) T1u (-3.1 eV) Hg (-8.2 eV) Energy Gg Gu Negatives of ionization potentials Hu (-7.1eV) for valence levels, calculated IP's and EAs are in good agreement with experiment
Ab initio search for a polarization bound anion state of C60. • The anion is not bound in the Hartree-Fock approximation, so cannot use approaches that assume Hartree-Fock provides a good starting point. • Instead we adopted the EOM-CCSD method, with a large flexible basis set of Gaussian functions The calculations predict the s-type anion to be bound by about 130 meV, Integrated probability ψ2 Ψ2*r2 30 5 15 25 35 5 10 15 10 20 40 20 R (Bohrs) 25 30 R (Bohrs) About 9% of the charge is located inside the C60 based on analysis of the dominant natural orbital for the excess electron
Occ. number of s-type natural orbital 0.985 • Several "filled" natural orbitals have occupations of about 1.9 and several empty natural orbitals have occupations 0.05 – 0.10 • These describe dispersion interactions between excess e- and the electrons of C60 • Dominant dispersion interactions when the excess e- is within ~ 4 Å of the C60 surface • About ~50 % of the excess electron density is further way • The long-range tail of the wavefunction of the excess electron is relatively unimportant for the dispersion interactions Unlike the (H2O)n clusters, ADC(2) overbinds the anion of C60 by ~2X (compared to EOM-CCSD): lack of screening? EOM-MP2 underbinds by about 40%. High-order correlation effects are more important for C60 than for the water clusters.
Electrostatic and polarization potentials for C60. Shaded area indicates the size of the C atoms as given by vdW radii Fit to the short range electrostatic and polarization potentials The polarization potential is not r-4 except at very large distances. Radial distribution Charge distribution is similar to that from the EOM-CCSD calculations Model potential Repulsive potential at the C60 radius builds in orthogonality
We are now working on developing a one-electron model Hamiltonian for describing polarization bound anions of C60, aggregates of C60, and other fullerenes • Here the challenge is to account for the charge-flow component of the polarizability of C60 +0.03 E_field = 0.001 a.u. Induced Dipole moment = 0.49 a.u. Charge range -0.03 A dipole moment of 0.49 a.u (1.2 D) is developed in a field of 0.001 a.u. in the +x direction. Much of this is due to charge-flow.
STM measurements of C6F6 on Cu(110) also displays electron capture into an extended orbital (Petek et al.) • Has been interpreted in terms of e- capture into valence σ* • Our work suggests that a non-valence correlation bound anion may be responsible • It is well known that C6F6 has a bound valence anion with a buckled geometry • EOM calculations bind the e- for both the planar and buckled structures • but give very different charge distributions for the two structures • Clearly non-valence in the planar structure
Electrostatic and polarization potentials of C6F6 in the z-direction (perpendicular to the molecule) Both the polarization and the electrostatic potentials are essential for the binding of the e- to the planar molecule The quadrupole moment is of opposite sign in benzene, and as a result, it does not have a quadrupole-bound anion
Diabatic and adiabatic states of C6F6- along the buckling coordinate There is an avoided crossing between the valence and non-valence diabatic anion states
CO2- shares a lot of characteristics with C6F6- The anion is valence in nature for highly bent structures (OCO angle < 148 deg), and is very extended for larger angles Neutral When using a large basis set, e.g., ANO +3s3p on each atom, the anion potential bends over for angles > 150 deg, when using methods that do not depend on the suitability of HF as a starting wavefunction. First elucidated by Sommerfeld et al.
Just beyond the crossing point of the neutral and anionic HF potentials, one can find two HF solutions to for the anion: • One with the excess electron localized and the other with it collapsed onto the continuum • The figure to the left depicts the potentials for the latter case. • Note that with a large basis set, the bending potential of CO2- does NOT correlate with the π* anion of the linear molecule • Walsh's rule breaks down • Has been discussed in papers by McCurdy and Rescigno
Correlation bound anions of Xen clusters Of interest since correlation effects dominate the binding (electrostatics of little importance) Results for EOM calculations. ADC(2) overbinds by up to 3x Is problem screening, or breakdown of use of uncoupled "HF" polarizabilities Xe20 and C60 have similar polarizabilities and similar EBEs when electrostatics suppressed in the latter
PISCES (Pittsburgh InfraStructure for Clusters with excess ElectronS) http://www.pisces.pitt.edu/ • PISCES is a code for describing the interaction of excess electrons with atomic and molecular clusters. It uses of a model Hamiltonian so that only the excess electron is treated explicitly. (Developed with the support from NSF) • Release 1.0: • Characterizes excess electrons interacting with water clusters using a DVR basis set • Polarizable DPP force-field for water • Self-consistent electron-water polarization with gradients • Ground and electronically excited states • Readily coupled with molecular dynamics and pathway searching codes • Planned Additions: • Excess electron states of inert gas atoms and fullerenes • Drude oscillator treatment of water molecules • Periodic boundary conditions
Summary • Molecules or clusters with sufficiently high polarizability will have non-valence correlation bound anions • These are closely related to the image potential states of metals and graphene • If the polarizability is not sufficiently high, the balance can be tipped by favorable electrostatics • In general EOM-MP2 is adequate for such anions (i.e, gives results close to EOM-CCSD) • One can develop one-electron model Hamiltonian approaches that accurately describe these non-valence ions
Tetramethyleneethane (TME) Non-kekule, disjoint diradical 6 π electrons with orbitals 3 and 4 being essentially degenerate Considerable debate in the literature as to the spacing between the lowest singlet and triplet states
A major complication, is that the molecule can rotate relatively freely about the central CC bond • In our DMC calculations we use the dominant configurations from CASSCF(6,6) calculations on the singlet and triplet states • About 25 determinants for each state • CI coefficients optimized together with the Jastrow factors
TME twisting potentials For the singlet state, the DMC potential has a rather different shape than the corresponding cas(6,6) potential CAS(6,6)PT2 results very similar to DMC if cc-pVTZ or better basis set is used CAS CASPT2 DMC
Two shortcomings of earlier work on TME • 1. Basis sets lacked f functions on C atoms • 2. A two-configuration reference space is inadequate for CASPT2 or MRCC • The energy gap between the first two π orbitals and the 2nd pair of π is not very large