Download

1 / 63
630 likes | 781 Vues
Modeling Splice Site and Transcription Factor Binding Site Variation by Information Theory. Peter K. Rogan, Ph.D. St. Jude’s Children’s Research Hospital Memphis, TN May 15, 2003. Background.

E N D
Modeling Splice Site and Transcription Factor Binding Site Variation by Information Theory Peter K. Rogan, Ph.D. St. Jude’s Children’s Research Hospital Memphis, TN May 15, 2003
Background • Information theory provides general solutions to the problem of how to recognize members of a group of related nucleic acid (or protein) sequences.
The average information of a related set of sequences, Rsequence, represents the total sequence conservation: Rsequence =2 - [ -f(b,l) log2f(b,l) + e(n(l)) ] f(b,l) is the frequency of each base b at position l, e(n(l)) is a correction for the small sample size n at position l Schneider et al. J. Mol. Biol. 1984
Sequence Logo Conservation and diversity among related binding sites can be visualized using a sequence logo. The area under the logo is Rsequence, the average Information of the binding site.
Definition of Individual Information • The individual information, Ri, of a single member of a sequence family is the dot product of that sequence vector and a weight matrix, Ri(b,l), based on the of the base frequencies at each position of the sequence. t Ri(j) = s(b,l,j) Riw(b,l) (bits per site j) l b=a
Distribution of Individual Information for related binding sites The average of the set of Ri values for a family of sequences isRsequence.
Second law of thermodynamics -kBT ln 2 q / R q: heat dissipated; T: temperature; R: information q < 0 => R > 0 q > 0 => R < 0 DNA Mutation or Unrelated sequence HLH Protein HLH Protein bound to WT DNA
Sequence Walker Definition Among related sequences having a common function, functional sites can be distinguished from non-sites with the sequence walker. (E. coli Fis protein) bits 2 0 -4
Sequence Walker Application I The matrix can be scanned along a “test sequence” until... bits 2 0 -4 Ri = - 6.7 bits at position 179 of the sequence. The Z score is -5.4.
Sequence Walker Application II … a greenbar indicates a potential binding site bits 2 0 -4 Ri = 9.2 bits at position 180 of the sequence. The Z score is 0.3.
mRNA splicing gene 5’ IVS 1 IVS 2 DNA 2 3 acceptor 1 3’ Exons donor Transcription IVS1 5’ hnRNA IVS2 3’ 2 3 1 Splicing or 1 1 2 3 3 Mature mRNA Alternative mRNA
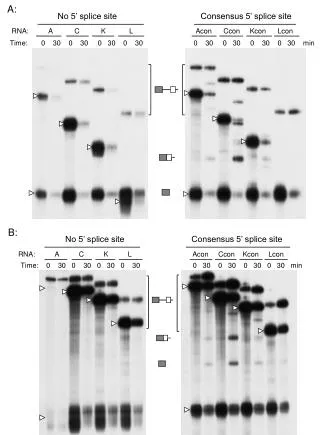
Splice Site Model Building • We extracted coordinates of unique donor and acceptor splice sites of known genes from the given strand of the 10/7/00 Human Genome Working Draft. • Valid splice junctions were evaluated by information theory (Ri > 0) and the Ri(b,l) matrix was computed. • This process was iterated (~ 10 cycles) until all sites evaluated with the matrix had Ri > 0.
Semi-automated Splice site Model Refinement • ~ 1/3 of exon-intron junctions are misaligned in the draft, • owing to the rapid alignment procedures used (ie. BLAT).
Ri analysis of sequence variation at binding sites • Effects of mutations • Effects of polymorphisms • Detection of cryptic sites • Relationship between information content and phenotype
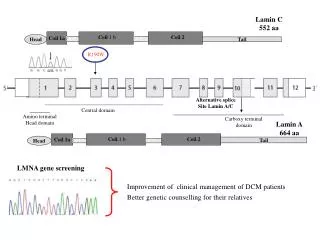
mRNA splicing mutations (*, ^) IVS 1 gene 5’ IVS 2 * * DNA 2 3 Exons acceptor 1 3’ donor IVS1 5’ hnRNA IVS2 * * ^ 3’ 3 1 2 ^ or or 1 2 3 1 2 3 1 3 Leaky or no wild type mRNA Exon skipping (*) Cryptic splicing (^) Mutant forms
The minimum information required for donor site recognition Temperature sensitive mutation in COL3A1 results in 50% exon skipping and Ehlers-Danlos syndrome, Type VII. Splicing is impaired at 39 deg.C and restored at 30 deg. C, which is consistent with weak binding by U1 splicesome.
Cryptic splicing mutations A C->T mutation in exon 3 of the iduronidate synthetase gene activates a cryptic donor site upstream of the natural donor site.
Mechanism of exon recognition U1 splicesome U2 splice + U2AF exon acceptor donor 3’ 5’ mRNA Binding sites
Mechanism of exon recognition: cryptic splicing mutation (2a) U1 splicesome U2 splice + U2AF exon Natural acceptor 3’ Natural donor 5’ mRNA Activated cryptic donor Either not recognized or to lesser degree Recognized Binding sites
CFTR Polymorphism (5T, 7T, 9T) Pop Freq 60% 35% 5% Splicing among 3 common alleles that differ in length in the polymorphic polythymidine tract of the IVS 8 acceptor of the CFTR gene.The shortest allele (top walker) shows 90% skipping of exon 9 and is associated with congenital absence of the vas deferens. Individuals with the two longer alleles have a normal phenotype, although the 7T allele produces less mRNA than the 9T allele.
Prediction of clinical phenotypes • Hereditary non-polyposis colon cancer • Hemophilia A and B • Atherosclerosis
Predicting Phenotype of HNPCC Splicing Mutations by Information Analysis Lynch II mutations Lynch I mutations
Results are consistent with MSH2 -/- and MSH2 +/- transgenic mouse phenotypes. Increased proliferation induces widespread DNA replication errors, which are repair normally until DNA repair systems are saturated (Cancer Res. 62:2092, 2002). Mismatch repair machinery is activated by DNA damaging agents (Nature 399:806, 1999; PNAS 96:10704, 1999).
Relating Information Content of F8C and F9 Splicing Mutations and Bleeding Phenotypes To predict severity of hemophilia, mutations in the factor VIII ( F8C ) or factor IX ( F9 ) genes were analyzed for changes in R : I v The receiver operating curve discriminated mildly or moderately from severely ³ 2.4 reduced protein activity for values D bits or R < 7 bits ( P =.001). i v Using these thresholds: - 91% of mutations with severely reduced protein expression were correctly identified (n=45; P< 0.001). - 86% of mutations associated with severe bleeding and all mutations with moderate bleeding symptoms were correctly identified (n= 22 p< .0009).
Information Content of Splicing Mutations in Lipid Metabolizing Genes vs. Phenotype Ri value cutoff (bits) Phenotype* Dyslipidemia Reduction in protein level or activity Mild Average Severe Mild Average Severe < 2.4 0/15 10/15 5/15 1/9 7/9 1/9 > 2.4 2/5 3/5 0/5 2/3 1/3 0/3 Fraction is the number of mutations in category / total number above or below 2.4 bits. Mutant genes included APOAII,APOB,APOCII,APOE,CBS,CETP,LCAT,LIPA,LDLR, and LPL.
Generating information models of eukaryotic transcription factor cis-regulatory binding sites • Unique challenges: • Variant sequences are not obvious • Requires experimental determination and validation • Effect of ascertainment bias • in published sites • in SELEX-generated sites • Binding protein does not necessarily signify that it activates (or represses) transcription
Greek Hereditary Persistence of Fetal Hemoglobin(HBGA, -119G>A) 6.8 bits 7.3 bits
The Transcription Factor Binding Site Problem: Bias in Models Derived from TRANSFAC data towards Consensus Sequences* *Consensus sequences have the strongest binding, but are often not representative of the majority of sites.
bits bits bits bits bits bits bits Refinement of the Pregnane X Receptor (PXR/RXRα) binding site model Initial PXR/RXR Model. Published PXR/RXR binding sites (n=15; and flanking sequences) were multiply aligned by minimization of uncertainty. The -2 to +20 interval contained most of the information, was consistent with published binding studies, and was therefore used to define the site.
Competition Curves for Novel PXREs Identified by Model 1 To quantify the relative affinity of PXR/RXR, band density was plotted versus pmol competitor to determine the concentration of competitor required to deplete PXR/RXRα binding to the CYP3A4 proximal PXRE by 50%. Relative binding was normalized to the band intensity of the reactions with no added competitor as 100%.
Comparison of predicted and measured binding affinities for novel PXR/RXRα sites identifiedwith the initial model Predicted fold differences in binding were closer to densitometrically-determined differences when these weaker sites were added in Model 2.
Model 2 Characteristics. (A) Alignment of PXREs derived from published literature and plus four new PXREs indicated in above Table (B) Histogram of strengths of sites in Model 2 versus frequency (C) Sequence logo of PXR/RXR binding site. Ri values are more Gaussian distributed than the initial model. (B) (A) (C) Model 2 Characteristics • Alignment of published + validated PXREs • (B) Histogram (C) Sequence logo Model 2 Characteristics. (A) Alignment of PXREs derived from published literature and plus four new PXREs indicated in above Table (B) Histogram of strengths of sites in Model 2 versus frequency (C) Sequence logo of PXR/RXR binding site. Ri values are more Gaussian distributed than the initial model.
dNR1 dNR1 Proximal PXRE Proximal PXRE dNR2 dNR2 dNR3 dNR3 Ri Ri Scans of CYP3A4 and CYP2B6 promoters Each promoter was scanned with PXR/RXR model 2. Ri values are plotted versus the position of the PXRE in the CYP3A4 gene or the CYP2B6 gene. Ri values of sites on the antisense strand are shown upside down. Previously characterized PXR binding sites identified by the model are indicated in color.
Activation of the CYP2B6 Distal PXRE Transient transfections with CYP2B6 and control CYP3A4 PXRE fusion constructs. Rifampin induced luciferase activitiy 4- to 5-fold in cells cotransfected with an expression plasmid for human PXR and CYP2B6-dPXRE(2X)-luc, and 2- to 3- fold in cells cotransfected with CYP3A4-pPXRE(2X)-luc. Rifampin had no effect on luciferase activity in cells transfected with the enhancerless-reporter. Average luciferase activity ± SD of three replicates from 3 independent transfections is shown.
PXR/RXR Model 3 Weaker binding sites from well established PXR/RXRα target gene promoters (Ri < Rsequence) were validated and introduced into Model 3.
Novel validated binding sites in Model 4 These 14 binding sites are not present in the Nov 02 human genome draft!
Possible significance of novel sites • Not present in reference sequence, but they are polymorphisms or mild mutations • Advantage is that binding is not abrogated, but reduced, ie. gene is less PXR/RXR responsive. • Possible “wobble” code for regulatory elements • Ancestral binding sequence present in primate lineage • PXR/RXR mutation rate is slower than cis-regulatory element; protein retains ability to recognize sequences that are no longer present • This could explain why heterologous cross-species transfections are faithfully regulated.
Development of a Xenobiotic biosensor based on the information theory-derived optimal site HepG2 cells were transiently transfected with 100 ng luciferase reporter, 5 ng pRL-CMV and 25 ng pSG5-hPXRDATG with Lipofectamine Plus. After treatment for 24 hours with 10 mM Rifampin or 0.1% DMSO (solvent), cells were harvested and Dual-luciferase assays were performed. Results are the average of three separate wells transfected and treated in parallel.
Histogram of binding site strengths for sites in genome scan >10 bits
Visualization of successive genome scans of PXR/RXRα binding site models
Monitoring PXR/RXR refinement through complete genome promoter scans