Schedule change
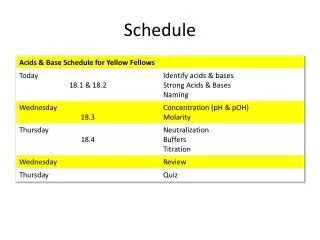
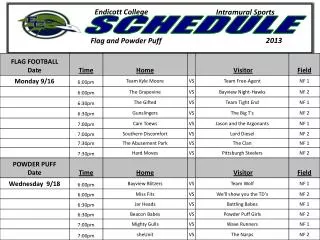
Schedule change . Galaxy server going down for maintenance on Thursday. Day 2: AM - Introduction to RNA-Seq (and a touch of miRNA-Seq) Day 2: PM - RNA-Seq practical ( Tophat + Cuffdiff pipeline on Galaxy) Day 3: AM – Introduction to Exome Sequencing and Variant Discovery


Schedule change
E N D
Presentation Transcript
Schedule change Galaxy server going down for maintenance on Thursday • Day 2: AM - Introduction to RNA-Seq (and a touch of miRNA-Seq) • Day 2: PM - RNA-Seq practical (Tophat + Cuffdiff pipelineon Galaxy) • Day 3: AM –Introduction to Exome Sequencing and Variant Discovery • Day 3: PM - Exome sequence analysis practical (Galaxy)
Quick Recap • NGS data production becoming commonplace • Many applications -> research intent determines technology platform choice • High volume data BUT error prone • FASTQ is accepted format standard • Must assess quality scores before proceeding • ‘Bad’ data can be rescued
The Central Dogma of Molecular Biology Reverse Transcription
RNAseq Protocols • cDNA, not RNA sequencing • Types of libraries available: • Total RNA sequencing (not advised) • polyA+ RNA sequencing • Small RNA sequencing (specific size range targeted)
Genome-scale Applications • Transcriptome analysis • Identifying new transcribed regions • Expression profiling • Resequencing to find genetic polymorphisms: • SNPs, micro-indels • CNVs • Question: Why even bother with exome sequencing then?
Sequencing details • Standard sequencing • polyA/total RNA • Size selection • Primers and adapters • Single- and paired-end sequencing • Strand-specific sequencing • still immature tech • Sequencing only + or – strand • Mostly paired-end
What about microarrays??!!! • Assumes we know all transcribed regions and that spliceforms are not important • Cannot find anything novel • BUT may be the best choice depending on QUESTION
Arrays vs RNAseq (1) • Correlation of fold change between arrays and RNAseq is similar to correlation between array platforms (0.73) • Technical replicates almost identical • Extra analysis: prediction of alternative splicing, SNPs • Low- and high-expressed genes do not match
RNA-Seq promises/pitfalls • can reveal in a single assay: • new genes • splice variants • quantify genome-wide gene expression • BUT • Data is voluminous and complex • Need scalable, fast and mathematically principled analysis software and LOTS of computing resources
Experimental considerations • Comparative conditions must make biological sense • Biological replicates are always better than technical ones • Aim for at least 3 replicates per condition • ISOLATE the target mRNA species you are after • NOT looking for new transcripts can bias expression estimates
Analysis strategies • De novo assembly of transcripts: + re-constructs actual spliced transcripts + does not require genome sequence easier to work post-transcriptional modifications - requires huge computational resources (RAM) - low sensitivity: hard to capture low abundance transcripts • Alignment to the genome => Transcript assembly + computationally feasible + high sensitivity + easier to annotate using genomic annotations - need to take special care of splice junctions
Basic analysis flowchart Illumina reads Remove artifacts AAA..., ...N... Clip adapters (small RNA) "Collapse" identical reads Align to the genome Pre-filter: low complexity synthetic Count and discard Re-align with different number of mismatches etc un-mapped mapped mapped un-mapped Assemble: contigs (exons) + connectivity Filter out low confidence contigs (singletons) Annotate
Software • Short reads aligners • Stampy, BWA, Novoalign, Bowtie, TOPHAT • Data preprocessing • Fastx toolkit • samtools • Expression studies • Cufflinks package • R packages (DESeq, edgeR, more…) • Alternative splicing • Cufflinks • Augustus
The ‘Tuxedo’ protocol • TOPHAT + CUFFLINKS • TopHat aligns reads to genome and discovers splice sites • Cufflinks predicts transcripts present in dataset • Cuffdiff identifies differential expression Very widely adopted suite
‘Tuxedo’ protocol limitations • Uses shortread data - Illumina OR SOLiD • Requires a sequenced genome • No GUI • Versions implemented in GALAXY are old(ish)
Splice junctions • In humans, terminal exons are ~1kb long, and since mRNAs are ~2kb, • ~half of the reads should originate in initial and internal exons • Initial and internal exons are ~200b long • => for 75-mer reads, ~20% of reads are supposed to cross splice junctions R RNA: Lexon Genome:
Splice junctions strategies • Create a splice junctions database joining together donors and acceptors • Typically, use known (annotated) splice junctions or known splice sites • TopHat: uses putative exons from mapped reads, database is made of canonical splice sites around putative exons
Read alignment with TopHat (2) • Uses BOWTIE aligner to align reads to genome • BOWTIE cannot deal with large gaps (introns) • Tophat segments reads that remain unaligned • Smaller segments mostly end up aligning
Read alignment with TopHat (3) • When there is a large gap between segments of same read -> probable INTRON • Tophat uses this to build an index of probable splice sites • Allows accurate measurement of spliceform expression • Possibility of detecting gene fusion events
Cufflinks package • http://cufflinks.cbcb.umd.edu/ • Cufflinks: • Expression values calculation • Transcripts de novo assembly • Cuffcompare: • Transcripts comparison (de novo/genome annotation) • Cuffdiff: • Differential expression analysis
Cufflinks: Transcript assembly • Assembles individual transcripts based on aligned reads • Infers likely spliceforms of each gene • Builds ‘transfrags’ • The smallest number of spliceforms that can be explained by the data • NOTE:assembly errors do occur -> sequencing depthhelps
Cufflinks: Transcript assembly (2) • Quantifies expression level of each transfrag • Filters out those likely to be premature terminations, non-mature mRNAs, etc
Cuffmerge • Merges transfrags into transcripts where appropriate • Also performs a reference based assembly of transcripts using known transcripts • Produces single annotation file which aids downstream analysis
Cuffdiff: Differential expression • Calculates expression level in two or more samples • Expression level relates to read abundance • Because of bias sources, cuffdiff tries to model the variance in its significance calculation What else is important?
FPKM (RPKM): Expression Values • Fragments Reads Per Kilobase of exon model per Million mapped fragments • Nat Methods. 2008, Mapping and quantifying mammalian transcriptomes by RNA-Seq. Mortazavi A et al. C= the number of reads mapped onto the gene's exons N= total number of reads in the experiment L= the sum of the exons in base pairs.
Cufflinks (Expression analysis) gene_id bundle_id chr left right FPKM FPKM_conf_lo FPKM_conf_hi status ENSG00000236743 31390 chr1 459655 461954 0 0 0 OK ENSG00000248149 31391 chr1 465693 688071 787.12 731.009 843.232 OK ENSG00000236679 31391 chr1 470906 471368 0 0 0 OK ENSG00000231709 31391 chr1 521368 523833 0 0 0 OK ENSG00000235146 31391 chr1 523008 530148 0 0 0 OK ENSG00000239664 31391 chr1 529832 532878 0 0 0 OK ENSG00000230021 31391 chr1 536815 659930 2.53932 0 5.72637 OK ENSG00000229376 31391 chr1 657464 660287 0 0 0 OK ENSG00000223659 31391 chr1 562756 564390 0 0 0 OK ENSG00000225972 31391 chr1 564441 564813 96.9279 77.2375 116.618 OK ENSG00000243329 31391 chr1 564878 564950 0 0 0 OK ENSG00000240155 31391 chr1 564951 565019 0 0 0 OK
Cuffdiff (differential expression) • Pairwise or time series comparison • Normal distribution of read counts • Fisher’s test test_id gene locus sample_1 sample_2 status value_1 value_2 ln(fold_change) test_stat p_value significant ENSG00000000003 TSPAN6 chrX:99883666-99894988 q1 q2 NOTEST 0 0 0 0 1 no ENSG00000000005 TNMD chrX:99839798-99854882 q1 q2 NOTEST 0 0 0 0 1 no ENSG00000000419 DPM1 chr20:49551403-49575092 q1 q2 NOTEST 15.0775 23.8627 0.459116 -1.39556 0.162848 no ENSG00000000457 SCYL3 chr1:169631244-169863408 q1 q2 OK 32.5626 16.5208 -0.678541 15.8186 0 yes
Visualization: Genome Viewers • Web based: • UCSC Genome Browser (http://genome.ucsc.edu/) • Standalone • Integrated Genome Viewer (http://www.broadinstitute.org/software/igv/)
RNAseq hands-on practical (Galaxy) • Data QC and trimming • Aligning reads to reference genome • Running CUFFLINKS and looking at some transcripts using the UCSC genome browser • Finding differentially expressed genes with CUFFDIFF