Endocrine Hypertension
Endocrine Hypertension. Essential hypertension 92-94% Secondary hypertension 6-8% Renal 4-5% Miscellaneous ~2% Endocrine 1-2% Primary hyperaldosteronism 0.3-15% Cushing ’ s syndrome <0.1% Pheochromocytoma <0.1%.


Endocrine Hypertension
E N D
Presentation Transcript
Endocrine Hypertension Essential hypertension 92-94% Secondary hypertension 6-8% Renal 4-5% Miscellaneous ~2% Endocrine 1-2% Primary hyperaldosteronism 0.3-15% Cushing’s syndrome <0.1% Pheochromocytoma <0.1%
Endocrine Hypertension Mineralocorticoid excess Cushing’s syn. Pheochromocytoma
Epinephrine Norepinephrine Tyrosine Hydroxylase COMT AADC Metanephrine Normetanephrine Dopamine β Hydroxylase MAO PNMT Vanylmandelic Acid (VMA) CA Synthesis & Metabolism APUD cells
Pheochromocytoma • ~ 0.1% or less of hypertension • Extraadrenal- Paraganglioma “10% Tumor” Bilateral Extra-adrenal Familial(? ~20%) Malignant
Pheochromocytoma Symptoms During paroxysm:Between attacks: Headache Sweating Sweating Cold hands & feet Palpitations Weight loss Tremor Constipation Chest pain Abdominal pain Nausea, vomiting
Pheochromocytoma Signs: • Increased blood pressure • Orthostatic hypotension • Tachycardia
Pheochromocytoma Familial forms: (germ-line mutations autosomal dominant) Syndromic- Multiple Endocrine Neoplasia Type 2 (A & B) Multiple Endocrine Neoplasia Type 1 (rarely) von Hippel-Lindau disease Neurocutaneous syndromes (NF1) Non- syndromic SDHB, C, D; TEM127
When to Suspect a Pheochromocytoma? • Episodic HTN accompanied by the classical triad • Refractory HTN • Labile HTN • Severe pressor response to surgery etc. • Familial Hx associated with Pheo • Incidental adrenal mass • HTN at a young age • Takotsubo cardiomyopathy
MEN2 MEN 2A MEN 2B Familial Medullary Thyroid Carcinoma (FMTC) קרצינומה מדולארית של בלוטת התריס פאוכרומוציטומה היפרפרהטירוידיזם 10-20% 40-50% 90% 40-50% 100% 90%
MEN2b מראה מרפנואידי נוירומות בריריות
Von-HippelLindau Disease • Renal cell carcinoma • Retinal angioma • Cerebellar or spinal hemangioblastoma • Pheochromocytoma-7-19%.
Pheochromocytoma-Biochemical Diagnosis • 24 hour urine collection for free CATs • epinephrine and norepinephrine • Sensitivity ~70% • 24 hour urine collection for CAT metabolites (METS) • metanephrine, normetanephrine and acid • More specific, sensitivity >90% • Urinary CATS + METS • Sensitivity > 95% • Plasma free metanephrines. • Highest sensitivity > 95%
Pheochromocytoma imaging Only after the Dx of pheo is biochemically confirmed!! • CT • MRI • I123MIBG • PET-18FDG • Octreoscan or 68Ga-DOTATATE-PET
Pheochromocytoma- Therapy Surgical resection of tumor- • If localized to adrenal- laparoscopic adrenalectomy Prior to surgery- • Hypertension: α-blockade (phenoxybenzamine, doxasocin, phentolamine) • Tachycardia- β-blockade (only after α blockade)
Kidney 11 β HSD type 2 cortisol cortisone ANDROSTENDIONE Biosynthesis and action of Aldosterone Glomerulosa
Renin-Angiotensin- Aldosterone Regulatory System ↑ACTH Sympathetic stimulation
Mineralocorticoid Excess Hyperaldosteronism Primary Secondary Apparent MC excess ↓PRA and ↓ PAC
Hyperaldosteronism DD: Primary-PRA Secondary- PRA 1. Renal Artery Stenosis(atherosclerosis, fibromuscular dysplasia). 2. Primary tumor of the JGA
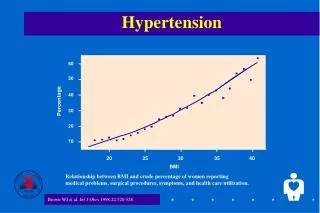
Primary Aldosteronism • Described by Conn in 1955. • Hypertension, hypokalemia, metabolic alkalosis. • Prevalence: 5-15% of patients with hypertension.
Causes of Primary Aldosteronism Aldosterone Producing Adenoma (APA) ~30% Idiopathic/hyperplasia (IHA) ~ 70% AdrenocorticalCarcinoma rare
Clinical Presentation Hypertension Moderate to severe (APA>hyperplasia) Refractory to medications K on low dose diuretics End-organ damage(aldosterone, HTN) LVH Micro and macro vascular disease
Clinical Presentation • Laboratory K, ↔K Metabolic alkalosis Mild Na • Symptoms related to hypokalemia Neuromuscular Nephrogenic DI (polyuria & nocturia)
Screening for Primary Hyperaldosteronism • Hypertension and hypokalemia(including patients treated with low dose diuretics). • Severe, resistant, or relatively acute hypertension, age<30. • An adrenal incidentaloma
Diagnosis: • Screening test • Confirmatory testing • Determine the subtype
Screening Tests for Primary Aldosteronism: PAC/PRA(PAC >20 ng/dl, PRA<1 ng/ml/h) • >30 suggestive; >50 diagnostic • Morning, ambulatory, paired, random PAC and PRA. • Serum K levels should be normalized • Not under beta blockers and spironolactone (preferably also w/o ACE inhibitors).
Diagnosis • Confirmatory tests: Saline infusion test-non suppressed aldo. 24 hour urinary aldosterone • Determining subtype: Posture test- normal response: elevation of Aldo IHA- normal response APA- no elevation
Imaging • Only after biochemical Dx(2-10% nonfunctioning adenomas on CT). • Abdominal spiral CT • In patients > 40 years of age-Selective adrenal venous sampling
Treatment • APA- Unilateral total adrenalectomy • IHA- Medical management (aldactone, amiloride, aplerenone)
Other Causes for Endocrine HTN • Hypothyroidism –diastolic HTN • Hyperthyroidism- systolic HTN • Acromegaly- salt retention • Hyperparathyroidism