
Uploaded by
olathe
0 SLIDES
459 VUES
0LIKES
Kinetics
DESCRIPTION
Kinetics. Ch 15. Kinetics. Thermodynamics and kinetics are not directly related Investigate the rest of the reaction coordinate Rate is important!. Chemical Kinetics. Kinetics – the study of the rates of chemical reactions
Download

1 / 0
Télécharger la présentation 
Kinetics
An Image/Link below is provided (as is) to download presentation
Download Policy: Content on the Website is provided to you AS IS for your information and personal use and may not be sold / licensed / shared on other websites without getting consent from its author.
Content is provided to you AS IS for your information and personal use only.
Download presentation by click this link.
While downloading, if for some reason you are not able to download a presentation, the publisher may have deleted the file from their server.
During download, if you can't get a presentation, the file might be deleted by the publisher.
E N D
Presentation Transcript
-
Kinetics
Ch 15 - Kinetics Thermodynamics and kinetics are not directly related Investigate the rest of the reaction coordinate Rate is important!
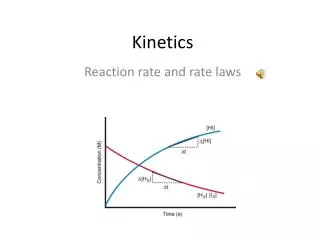
- Chemical Kinetics Kinetics – the study of the rates of chemical reactions Rate of reaction – change in concentration per unit time rate = Δ conc / Δ time Rate is generally not constant. It changes over the course of a reaction A B
- A B 10 18 24 28 31 33 What is happening to the rate of the reaction as time progresses? Why?
- Rate of Reaction A B Rate = Δ[B]/Δt = -Δ[A]/Δt Rate = Δ[product]/Δt = -Δ[reactant]/Δt
- Defining Rate Rate is defined arbitrarily by one pdt or rxt To be self consistent, Example: Stoichiometry important! 2 N2O5 (g) 4 NO2 (g) + O2 (g) Rate = Δ[O2]/ Δt 2 N2O5 (g) 4 NO2 (g) + O2 (g) Rate = Δ[NO2]/ 4Δt = - Δ[N2O5]/ 2Δt
- Another Example Calculated Rates Data
- Collect concentration data for reactants and products, then graph Effect of stoichiometry Average rate Instantaneous rate
- Rate Law Study rates to understand mechanism of reaction True rate depends on forward and reverse reactions (remember equilibrium?) But we can write rate law based on reactants Many reactions functionally irreversible Use initial rates (reverse rate is negligible)
- Rate Laws Two forms of rate law Differential Rate Law (Rate Law) How rate depends on concentration of reactants Experiment: Initial Rates of multiple trials Integrated Rate Law How concentrations of species depend on time Experiment: One trial sampled at multiple times
- Relationship Between Rate and Concentration 2 NO2 (g) + F2 (g) 2 NO2F (g) Rate = Δ[NO2F]/ 2Δt = -Δ[F2]/ Δt = -Δ[NO2]/ 2Δt Rate α [NO2] and [F2] Rate = k [NO2]x [F2]y k = rate constant x and y are the orders of reaction, these are determined experimentally – not from stoichiometry!
- 2 NO2 (g) + F2 (g) 2 NO2F (g) Rate = k [NO2]x [F2]y From experiment, x = 1 , y = 1 Rate = k [NO2] [F2] =Rate Law 1st order in NO2 , 1st order in F2, 2nd order overall One way to determine the rate law is from initial rates.
- H2O2 (aq) + 3 I- (aq) + 2 H+ (aq) I3- (aq) + 2 H2O (l) Rate = k[H2O2]x [I-]y [H+]z
- H2O2 (aq) + 3 I- (aq) + 2 H+ (aq) I3- (aq) + 2 H2O (l) Rate = k[H2O2]x [I-]y [H+]z Rate = k[H2O2][I-]
- Example Problem 2 NO2 (g) 2 NO (g) + O2 (g)
- Relationship Between Concentration and Time We want to use a single experiment to determine the rate law. We will do this by plotting concentration versus time. We will deal with simplest cases initially—only one reactant, generally “A”
- Zero Order Reaction How can a reaction rate be concentration independent?
- Zero Order Reactions Zero order reaction A B Rate = k[A]0 = k Rate = -d[A]/dt k = -d[A]/dt Rearrange and integrate from time = 0 to time = t [A]t – [A]o = -kt [A]t= -kt + [A]o
- Graphing Zero Order [A]t = -kt+ [A]o y = mx + b Plot of conc. vs. time gives straight line with slope of -k Units of k are M/s
- First Order Reactions Plot of concvs time does not give straight line (not 0 order) Rate changes over time: Doubling concentration of A doubles the rate A products Rate = k[A] Rate = -d[A]/ dt k[A] = -d[A]/ dt UTMOCln[A]/[A]0 = - kt ln[A] = -kt + ln[A]0
- Example Problem The decomposition of N2O5 to NO2 and O2 is first order with k = 4.80 x 10-4 s-1 at 45 oC. If the initial concentration is 1.65 x 10-2 M, what is the concentration after 825 sec? How long would it take for the concentration of N2O5 to decrease to 1.00 x 10-2 M?
- Graphing First Order Plot of ln[A] vs t gives straight line with a slope of -k and a y-intercept of ln[A]0 Units of k = s-1 2 N2O5 4 NO2 + O2
- First Order Reactions Determine Order of Reaction by plotting data!
- 0.27 0.24 0.18 2 N2O5 (g) 4 NO2 (g) + O2 (g)
- NOT Zero order!
- 0.14 0.13 0.14 0.13
- This is First Order
- Second Order Reactions Plot of [conc] vs t and plot of ln[conc] vs t do not give straight lines. (not 0 or 1st ) A products Rate = k[A]2 Rate = -Δ[A]/ Δt k[A]2 = -Δ[A]/ Δt UTMOC 1/[A] = kt + 1/[A]0
- Graphing Second Order Plot of 1/[A] vs t gives straight line, with a slope of k and a y-intercept of 1/[A]0
- 2 HI (g) H2 (g) + I2 (g) @ 580K
- Not Zero Order
- Not First Order
- Second Order Rate = k[HI]2
- Reactions Involving Gases A (g) products PV = nART [A] = nA / V = P/RT ln[A]/[A]0 = -kt ln(P/RT) /(P0/RT) = -kt ln P/P0 = -kt Can use the pressures of gases for the concentrations.
- Half Life k = describes speed of the reaction Large k = fast reaction Another way to describe speed is to use t½, the half life. This is the time needed to decrease to ½ [A]0. For a first order reaction, t = (1/k)ln[A]0/[A] t½ = (1/k) ln[A]0/([A]0/2) t½ = (1/k) ln2 = 0.693/k
- Comparison of Half-Lives Use same procedure to derive each half-life For zero order, each half life is half as long as previous one For first order, each half-life is the same For second order, each half life is twice as long as the previous one
- Application Question Kinetic data were plotted for A 2B + C What can these data tell you about this reaction?
- More Complicated System So far we have assumed one reactant How do we study A + 2B C + 3D Run experiment with B in huge excess Rate = k [A]x[B]y but [B] remains constant Called “psuedo” kinetics—can be used to determine order of A Repeat with excess A to get pseudo-order of B Combine experiments to get real rate law
- Reaction Mechanisms A reaction may be more complex that 1 simple collision – may form intermediates. It is unlikely that 3 or more molecules will collide simultaneously. Elementary steps – describe a molecular event.
- NO2 + CO NO + CO2 2 elementary steps NO2 + NO2 + NO3 + CO NO3 + NO + NO2 + CO2 NO2 + CO NO + CO2 NO3 is an intermediate
- Cl2 2 Cl Cl + CHCl3 HCl + CCl3 Cl + CCl3 CCl4 Cl2 + CHCl3 HCl + CCl4 (overall) Rate laws for elementary steps can be written from stoichiometry. (unlike overall) Rate = k1[Cl2] unimolecular Rate = k2[Cl][CHCl3] bimolecular Rate = k3[Cl][CCl3] bimolecular
- Rate Determining Step Rate Determining Step (rate limiting step) – is the slowest step leading to the formation of the products (slow step). The rates of any steps after the slow step are not important.
- O3 + 2 NO2 O2 + N2O5 O3 + NO2 NO3 + O2 slow NO3 + NO2 N2O5 fast Rate = k[O3][NO2] This explains why stoichiometry and rate law are independent
- 2 NO2 + O3 N2O5 + O2 Rate = k[NO2][O3] 2 NO + Cl2 2 NOCl Rate = k[NO]2[Cl2]
- Temperature Changes Rate 2NO (g) + Cl2 (g) 2NOCl (g) Rate = k[NO]2 [Cl2] k @ 25 oC = 4.9 x 10-6 M-1s-1 k @ 35 oC = 1.5 x 10-5 M-1s-1 This is more than 3x increase! Why is there a temperature dependence on k? Can k and temperature be related theoretically and quantitatively?
- Dependence of Rate Constant on Temperature Exponential of rate constant on absolute temperature Every curve different This one represents double rate every 10 K
- Collision Theory Collision Theory – molecules must collide in order to react. Rate α # collisions / sec α [reactants] From KMT Increase temp, increase speed Accounts for higher rate Kinetic energy made into potential energy to break bonds
- Activation Energy Arrhenius – expanded collision theory (1888) Molecules must collide with enough energy to rearrange bonds. If not, they just bounce off. Activation Energy = Ea = minimum amount of energy required to initiate a chemical reaction. Activated Complex (transition state) – temporary species in reaction sequence, least stable, highest energy, often undetectable.
- Activation Energy and Transition States
- Molecules with Enough KE to Overcome Activation Energy Boltzmann distribution Doubling temperature more than doubles fraction of molecules with enough energy Fraction =
- Steric Factor Rate constant k depends on three things: 1. Need to collide (z = collision rate) 2. Need E > Ea(fraction of collisions is 3. Need to be oriented in the right way to react! Steric factor (orientation factor) = p 0>p>1 k = zp k = A A is pre-exponential factor, or frequency factor, for the Arrhenius equation
- Orientation of Molecules
- Arrhenius Equation k = Ae-Ea/RT ln k = lnAe-Ea/RT y = m x + b If you experimentally determine ___ and ____, then you can graphically determine ____ and ____. Can also be used to determine k at any ____.
- ln k vs. 1/T m = -Ea/R b = ln A
- Useful Form of Arrhenius Equation In principle, best to do many experiments, graph line, and determine Ea In practice, can get decent value from only two experiments Rearrange Arrhenius equation to get
- Example Problem H2 + I2 2 HI k = 2.7 x 10-4 M-1s-1 @ 600 K k = 3.5 x 10-3 M-1s-1 @ 650 K a. Find Ea b. Calculate k @ 700 K
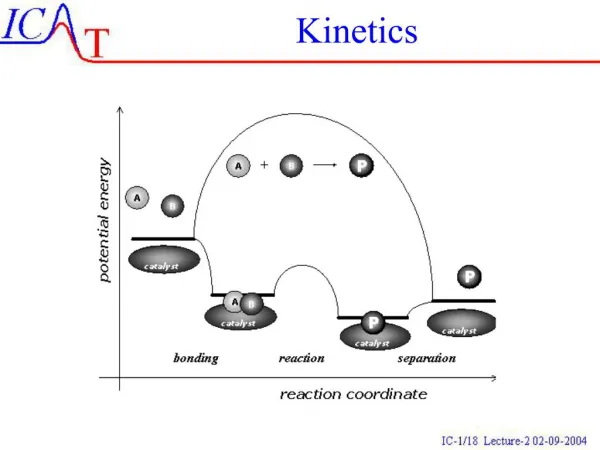
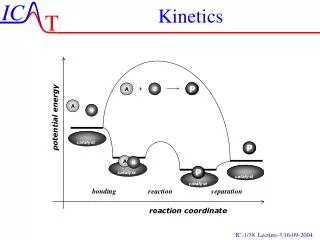
- Catalysis To increase the rate of a reaction 1. Increase temperature 2. Add a catalyst Catalyst – a substance which increases the rate of a reaction but is not consumed. Catalysts are involved in the course of a reaction. Usually by lowering Ea Homogeneous – catalyst the same phase as the reactants Heterogeneous – catalyst in different phase (usually a solid)
- Ammonia Formation with a Catalyst N2 + 3 H2 2 NH3
- Catalytic Hydrogenation Food processing Trans fatty acids



















More Related