Primary hyperlipidemias
Primary hyperlipidemias. Dr Shreetal Rajan , Senior Resident, Cardiology,MCH,Calicut. Primary hyperlipidemias. Classification of hyperlipidemias Overview on lipid metabolism Primary hyperlipidemias Management. Terminology. Hyperlipidemia


Primary hyperlipidemias
E N D
Presentation Transcript
Primary hyperlipidemias Dr Shreetal Rajan , Senior Resident, Cardiology,MCH,Calicut
Primary hyperlipidemias • Classification of hyperlipidemias • Overview on lipid metabolism • Primary hyperlipidemias • Management
Terminology Hyperlipidemia • Concentration of lipid in the blood exceeds the upper range of normal in a 12 hr fasting blood sample • Includes both hypercholesterolemia and hypertriglyceridemia Dyslipidemia • Dyslipidemia – derangement in blood lipid concentration or composition • Almost always due to hyperlipidemia • Dyslipidemia – major role in atherosclerosis and CAD
Lipoprotein structure • hydrophobic core • triglyceride and/or • cholesterol ester • surface coat • phospholipid monolayer • interspersed free cholesterol and apolipoproteins
The lipoprotein fractions • Chylomicrons • Very Low density lipoproteins (VLDL) • Intermediate density Lipoproteins (IDL) • Low density Lipoproteins (LDL) • High density Lipoproteins (HDL)
Lipoproteins – physiological functions • absorption of - dietary cholesterol - long-chain fatty acids - fat-soluble vitamins • transport of - triglycerides - cholesterol - fat-soluble vitamins - from the liver to peripheral tissues • transport of cholesterol - from peripheral tissues to the liver
Apolipoproteins - functions • proteins associated with lipoproteins. • lipoprotein assembly and function. • activate enzymes in lipoprotein metabolism. • ligands for cell surface receptors.
The story of lipids – the normal physiology • Chylomicrons transport fats from the intestinal mucosa to the liver • In the liver, the chylomicrons release triglycerides and some cholesterol and become low-density lipoproteins (LDL). • LDL then carries fat and cholesterol to the body’s cells. • High-density lipoproteins (HDL) carry fat and cholesterol back to the liver for excretion.
Why study of lipoproteins and apolipoproteins are important? • Atherosclerosis and dyslipoproteinemias have a very close association • All the cardiovascular risk models advocate lipoprotein studies in risk stratification and prognostication • Recently, non – HDL fraction, apo B , ratio of apo B to apo A 1, number and size of small, dense LDL particles are all emerging as risk markers for CAD. • Subendothelial retention of LDL -initiating factor for atherosclerotic plaque formation
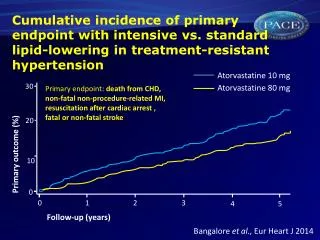
Attributable Risk Factors for a First Myocardial Infarction INTERHEART Study 100 90 80 60 50 PAR (%) 36 33 40 20 18 20 12 14 10 7 0 Hyper-tension Smoking Fruits/Veg Exercise Alcohol Abdominal obesity Psycho-social Lipids All 9 risk factors Diabetes Lifestyle factors n=15,152 patients and 14,820 controls in 52 countries MI=Myocardial infarction, PAR=Population attributable risk (adjusted for all risk factors) Source: Yusuf S et al. Lancet. 2004;364:937-952
Classification - hyperlipidemia • Primary • Secondary defect in genes and /or enzymes involved in lipoprotein metabolism 1st case report of Familial hypercholesterolemia • In 1938 Carl Mu¨ller, a Norwegian clinician, described FH as an “inborn error of metabolism” that produces high blood cholesterol and myocardial infarctions (heart attacks) in young people
Alternative classification I . Primary • Primary Disorders of Elevated ApoB -Containing Lipoproteins • Inherited Causes of Low Levels of ApoB -Containing Lipoproteins • Genetic Disorders of HDL Metabolism • Miscellaneous- Elevated Plasma Levels of Lipoprotein(a) Elevated small dense LDL particles II . Secondary forms of hyperlipidemia
Primary Disorders of Elevated Apo B -Containing Lipoproteins • Lipid disorders associated with elevated LDL and normal triglycerides • Lipid disorders associated with elevated triglycerides
Lipid disorders associated with elevated LDL and normal triglycerides • Familial Hypercholesterolemia (FH) • Familial Defective ApoB-100 (FDB) • Autosomal Dominant Hypercholesterolemia Due to Mutations in Pcsk9 (ADH-Pcsk9 or ADH3) • Autosomal Recessive Hypercholesterolemia (ARH) • Sitosterolemia • Polygenic Hypercholesterolemia
Familial hypercholesterolemia • Autosomal codominant disorder • Elevated plasma levels of LDL-C • Triglyceride level-normal • Premature coronary atherosclerosis Pathophysiology • Defect in LDL receptor • Homozygous and heterozygous • Receptor negative : < 2% LDL receptor activity • Receptor defective: 2-25% receptor activity
Familial hypercholesterolemia • tendonxanthomas –hands, wrists, elbows, knees, heels or buttocks • Total cholesterol levels > 500 mg/Dl • Accelerated atherosclerosis – begins in aortic root and extends into coronary ostia • Receptor negative-untreated patients don’t survive beyond 2nd decade • Receptor defective- better prognosis
Familial Defective Apob-100 (FDB) Dominantly inherited disorder Elevated plasma LDL levels with normal triglycerides, tendon xanthomas, increased incidence of premature ASCVD mutations in the LDL receptor–binding domain of apoB-100 LDL binds the receptor with reduced affinity -> removed from the circulation at a reduced rate Clinically identical to heterozygous FH but have lower plasma levels of LDL
Autosomal Dominant Hypercholesterolemia - physiology AD disorder ; gain-of-function mutations in PCSK9 PCSK9 is a secreted protein that binds to the LDL receptor causing its degradation LDL is internalized along with the receptor after binding In the low pH of the endosome LDL dissociates from the receptor and the receptor returns to the cell surface The LDL is delivered to the lysosome
Autosomal Dominant Hypercholesterolemia- pathology When PCSK9 binds to the receptor, the complex is internalized and the receptor is redirected to the lysosome rather than to the cell surface The missense mutations enhance the activity of PCSK9 The number of hepatic LDL receptors is reduced indistinguishable clinically from patients with FH
Autosomal Recessive Hypercholesterolemia (ARH) LDL Receptor Adaptor Protein (LDLRAP) is involved in LDL receptor–mediated endocytosis in the liver. In the absence of LDLRAP, lipoprotein-receptor complex fails to be internalized Hypercholesterolemia, tendon xanthomas, premature CAD Hyperlipidemia responds partially to treatment with HMG-CoA reductase inhibitors Usually require LDL apheresis to lower plasma LDL-C
Sitosterolemia Autosomal recessive disease severe hypercholesterolemia, tendon xanthomas, premature ASCVD (Atherosclerotic CardioVascular Disease) mutations in either of two members of the ATP-binding cassette (ABC) half transporter family, ABCG5 and ABCG8 genes are expressed in enterocytes and hepatocytes
Sitosterolemia • intestinal absorption of sterols is increased and biliary excretion of the sterols is reduced • increased plasma and tissue levels of both plant sterols and cholesterol • Dysmorphic red blood cells and megathrombocytes • hemolysis - distinctive clinical feature of this disease • respond to reductions in dietary cholesterol content • do not respond to statins. • Bile acid sequestrants and cholesterol absorption inhibitors - effective
Polygenic Hypercholesterolemia Elevated LDL with a normal plasma level of triglyceride in the absence of secondary causes of hypercholesterolemia Plasma LDL levels are generally not as elevated as they are in other primary hypercholesterolemias Family studies to differentiate polygenic hypercholesterolemia from single-gene disorders
Lipid Disorders Associated with Elevated Triglycerides • Familial Chylomicronemia Syndrome (Type I Hyperlipoproteinemia; Lipoprotein Lipase and ApoC-II Deficiency) • Familial Dysbetalipoproteinemia (Type III Hyperlipoproteinemia) • Apo A-V Deficiency • GPIHBP1 Deficiency • Hepatic Lipase Deficiency • Familial Hypertriglyceridemia (FHTG) • Familial Combined Hyperlipidemia (FCHL)
Familial Chylomicronemia Syndrome LPL (Lipoprotein Lipase) is required for the hydrolysis of triglycerides in chylomicrons and VLDLs apoC-II is a cofactor for LPL Genetic deficiency or inactivity of LPL or apo C II results in impaired lipolysis and elevations in plasma chylomicrons The fasting plasma is turbid Very high triglyceride levels
Familial Chylomicronemia Syndrome Present in childhood with features suggestive of acute pancreatitis Lipemia retinalis Eruptive xanthomas Hepatosplenomegaly Premature CHD not a feature
Familial ChylomicronemiaSyndrome- diagnosis IV heparin injection - endothelial-bound LPL is released LPL activity is profoundly reduced in both LPL and apo C-II deficiency normalizes after the addition of normal plasma (providing a source of apoC-II)
Familial Chylomicronemia Syndrome dietary fat restriction with fat-soluble vitamin supplementation medium-chain triglycerides Fish oils Fresh frozen plasma – source of apo C Plasmapheresis in pregnancy
HYPERTRIGLYCERIDEMIA - OTHER CAUSES APO A V DEFICIENCY • Apo A-V required for the association of VLDL and chylomicrons with LPL • Deficiency presents as hyperchylomicronemia GPIHBP1 Deficiency • LPL is attached to a protein on the endothelial surface of capillaries called GPIHBP1 • mutations that interfere with GPIHBP1 synthesis or folding cause severe hypertriglyceridemia
Hepatic Lipase Deficiency autosomal recessive disorder elevated plasma levels of cholesterol and triglycerides (mixed hyperlipidemia) due to the accumulation of circulating lipoprotein remnants association of this genetic defect with ASCVD is not clearly known Lipid-lowering therapy with statins along with other drugs
Familial Dysbetalipoproteinemia –FDBL (Type III Hyperlipoproteinemia) mixed hyperlipidemia; due to genetic variations in apoE Patients homozygous for the E2 allele (the E2/E2 genotype) comprise the most common subset of patients with FDBL precipitating factors usually present hyperlipidemia, xanthomas, premature coronary disease, peripheral vascular disease
Familial Dysbetalipoproteinemia (Type III Hyperlipoproteinemia) The disease seldom presents in women before menopause Two distinctive types of xanthomas- tuberoeruptive and palmar Broad beta band on electrophoresis Premature CHD Dramatic response to weight reduction and dietary changes; statins Treatment of other metabolic conditions
Familial Hypertriglyceridemia (FHTG) • The diagnosis of FHTG is suggested by the triad of • Elevated levels of plasma triglycerides (250–1000 mg/dL) • Normal or only mildly increased cholesterol levels (<250 mg/dL) • Reduced plasma levels of HDL-C • Plasma LDL-C levels are generally not increased and are often reduced due to defective metabolism of the triglyceride-rich particles
Familial Hypertriglyceridemia (FHTG) • type IV and type V of Fredrickson classification • autosomal dominant disorder of unknown etiology • VLDL is elevated • Precipitating factors • not associated with increased risk of ASCVD • secondary causes of hypertriglyceridemia to be ruled out • Monitor pancreatitis
Familial Combined Hyperlipidemia (FCHL) • autosomaldominant • one of three phenotypes • Elevated plasma levels of LDL-C • Elevated plasma levels of triglycerides due to elevation in VLDL • Elevated plasma levels of both LDL-C and triglyceride • classical feature of FCHL -lipoprotein profile can switch among these three phenotypes in the same individual over time • Associated with other metabolic risk factors • Family history of hyperlipidemia and/or premature CHD
Familial Combined Hyperlipidemia (FCHL) • significantly elevated plasma levels of apoB (Hyperapobetalipoproteinemia) • Increased small, dense LDL particles are characteristic of this syndrome • Overproduction of VLDL by liver – cause not known
Inherited Causes of Low Levels of Apo B Containing Lipoproteins Familial Hypobetalipoproteinemia (FHB) • MOST COMMON INHERITED FORM OF HYPOCHOLESTEROLEMIA • low total cholesterol and LDL-C due to mutations in apoB • LDL levels < 80 mg% • Protection from CHD • Parents have abnormal lipid fractions
Pcsk9 Deficiency • Loss of function mutations • PCSK9 normally promotes the degradation of the LDL receptor • Absence cause increased activity of LDL receptor and low LDL levels ( 40% reduction) • Protection from CHD increases as plasma LDL levels decrease
Abetalipoproteinemia • autosomal recessive disease • loss-of-function mutations in the gene encoding microsomal triglyceride transfer protein (MTP) • transfers lipids to nascent chylomicrons and VLDLs in the intestine and liver • Parents have normal lipid levels • diarrhea and failure to thrive • Neurologic manifestations • Pigmented retinopathydefective absorption and transport of fat soluble vitamins – vitamin E • low-fat, high-caloric, vitamin-enriched diet
Genetic Disorders of HDL Metabolism • Inherited causes of low levels of HDL-C • Gene Deletions in the Apo A V-AI-CIII-AIV Locus and Coding Mutations in ApoA-I • Tangier Disease (ABCA1 Deficiency) • LCAT Deficiency • Primary Hypoalphalipoproteinemia • Inherited causes of high levels of HDL-C • CETP Deficiency • Familial Hyperalphalipoproteinemia
Gene Deletions in the ApoAV-AI-CIII-AIV Locus and Coding Mutations in ApoA-I • Absence of mature HDL • Free cholesterol increase in HDL and in tissues • corneal opacities and planar xanthomas • Premature CHD
Tangier Disease (ABCA1 Deficiency) • autosomal recessive • ABCA1, a cellular transporter that facilitates efflux of unesterified cholesterol and phospholipids from cells to apoA-I • extremely low circulating plasma levels of HDL-C (<5 mg/dL) and apoA-I (<5 mg/dL). • hepatosplenomegaly , pathognomonic enlarged grayish yellow or orange tonsils, mononeuritis multiplex • Premature CHD not so common – because LDL levels also low
LCAT Deficiency • Autosomal recessive • defective formation of mature HDL 2 types – complete and partial Progressive corneal opacification Low levels of HDL COMPLETE FORM – hemolytic anemia, progressive renal insufficiency and ESRD PREMATURE CHD not seen
Primary Hypoalphalipoproteinemia (isolated low HDL Syndrome) • defined as a plasma HDL-C level below the tenth percentile in the setting of relatively normal cholesterol and triglyceride level • no apparent secondary causes of low plasma HDL-C • no clinical signs of LCAT deficiency or Tangier disease. • Premature CHD not a consistent feature
Inherited causes of high levels of HDL-C CETP DEFICIENCY • Loss-of-function mutations • CETP facilitates transfer of cholesteryl esters from HDL to apoB-containing lipoproteins • CETP deficiency results in an increase in the cholesteryl ester content of HDL,decreased clearance of HDL and a reduction in plasma levels of LDL-C • The relationship of CETP deficiency to ASCVD remains unresolved
Inherited causes of high levels of HDL-C Hyperalphalipoproteinemia • defined as a plasma HDL-C level above the ninetieth percentile • mutations in endothelial lipase • Relation to reduced CHD risk and increased longevity not consistent
Management-What are the recommendations? Checking lipids • Nonfasting lipid panel • measures HDL and total cholesterol • Fasting lipid panel • Measures HDL, total cholesterol and triglycerides • LDL cholesterol is calculated: • LDL cholesterol = total cholesterol – (HDL + triglycerides/5)