Download
1 / 1
30 likes | 148 Vues
This study explores the implementation of a lone-pair-based hydrogen-bonding energy function within the Mini-Rosetta framework. By developing a more accurate energy model that incorporates spatial constraints and couples parameters like distance and angles, we aim to improve the computational efficiency and visualization of hydrogen bonds. The approach is validated through Monte Carlo/gradient optimizations across a dataset of 28 protein structures, demonstrating how the new potential increases agreement with native structures from the PDB. Future work will refine the potential to include all hydrogen bond types and re-weight the hydrogen bond term relative to other potential energy contributions.

E N D
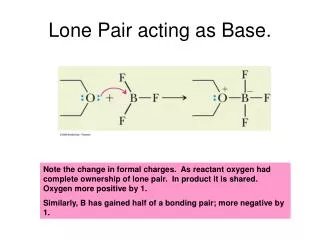
D H B A L2 L1 Computational Experiments with a Lone-Pair Based Hydrogen-Bonding Energy Function in Mini-Rosetta Alternative: Lone-Pair Energy Function Standard Hydrogen Bond Energy Function in Rosetta • Different Parameters involving hydrogen docking sites - “lone pairs” - can we have an in-plane constraint without • adding BAHD dihedral? (for computational efficiency, ease of visualization, etc.) • Remove the independence assumption (couple parameters) - distance and two angles. • Energy = F(AH, DHL, ALH ) • where A : acceptor atom, H : proton, D: donor, L : lone-pair (docking site) (Fig. 2) • • Fit polynomial to 3-dimensional energy surface • derived from database of 5200 protein structures. (Fig. 3) (starting with sp2 acceptor, side-chain hydrogen bonds) • Rosetta is an ab-initio protein modeling program • which includes a hydrogen-bond energy term. (Ref 1) • Energy is a function of molecular geometry. • Sum of independent terms (distance and two angles): Energy = F1(AH) + F2(BAH) + F3(AHD) where A : acceptor atom, B : base atom, H : proton, D : donor atom (Fig, 1) • BAHD dihedral (in-plane constraint) not implemented. • Hydrogen-bonds are divided into groups : helix, side-chain sp2, side-chain sp3, etc. and these groups are considered separately. Fig. 1: Hydrogen Bonding Parameters used by Rosetta AH distance, (BAH angle), (DHA angle). X(BAHD dihedral) not implemented. Fig. 2 : Parameters for Lone Pair energy function. AH distance, DHL angle, ALH angle. Fig 3: Histogram (isosurfaces) based on lone-pair parameters for the sp2 acceptor, side-chain hydrogen bonds for the 5200 protein database. Methods •Implement Lone-pair for sp2 acceptor side-chain hydrogen bonds. • Run Monte Carlo/gradient optimizations (“classic relax”) with lone-pair and standard potentials. • 28 protein set (60 - 300 residues each) • 10 relax runs for each protein. • Examine BAHD dihedrals in relaxed structures - compare with native structures from the PDB. BAHD dihedral : Lone-Pair Energy Structure Improves Agreement with PDB structures Fig. 5 : Histogram of BAHD dihedral for structures relaxed with the standard potential for the 26 protein set (10 relaxed structures per protein) (sp2 acceptor, side-chain hydrogen bonds) 8316 hydrogen bonds. Fig. 6 : Histogram of BAHD dihedral for structures relaxed with the lone-pair potential for the 26 protein set (10 relaxed structures per protein) (sp2 acceptor, side-chain hydrogen bonds) 5738 hydrogen bonds. Fig. 4 : Histogram of BAHD dihedral for the 5200 protein set (native PDB structures). Note peak centered at 0°. (sp2 acceptor, side-chain hydrogen bond.) 125261 hydrogen bonds Figure 2: Gary Bishop giving a head-mounted display demo to UNC System President Molly Broad. (Arial 24 pt italic) Future Directions Extend potential to all hydrogen-bond types. Re-weight hydrogen-bond term relative to other potentials. Predict structures with new potential, compare to natives and to predicted structures developed using standard. Acknowledgements : Kuhlman lab - Department of Biochemistry, UNC-CH Richardson lab - Duke University HERE Christopher Sheldahl Snoeyink Lab Department of Computer Science, UNC-CH YOUR NAME HERE (Arial 28 pt italic) YOUR PROJECT URL HERE